

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Chronic rhinosinusitis (CRS) is characterized by persistent inflammation of the nasal and paranasal sinus mucosa. Clinical manifestations of CRS include persistent rhinorrhea, nasal congestion, hyposmia, and/or facial pressure or pain that persists for more than 12 weeks [1]. CRS can be categorized into two groups: chronic rhinosinusitis with nasal polyps (CRSwNP) and chronic rhinosinusitis without nasal polyps (CRSsNP). The underlying mechanisms of CRS are complex, involving multiple pathological features such as frequent damage to the mucosal epithelium, thickening of the subepithelial basement membrane, gland hyperplasia, increased deposition of the extracellular matrix, and infiltration of inflammatory cells [2]. The process of epithelial-to-mesenchymal transition (EMT), in which epithelial cells lose their junctions and apical-basal polarity and acquire mesenchymal properties, including motility and the capacity for migration [3], has been implicated in the development of CRSwNP. Nasal epithelium derived from patients with CRSwNP exhibits decreased expression of E-cadherin and β-catenin (epithelial markers) and increased expression of vimentin and α-smooth muscle actin (α-SMA; mesenchymal markers) at both the mRNA and protein levels [2,4]. Additionally, EMT-inducing transcription factors such as Twist, Zeb, and Snail are also expressed at higher levels [5].
Tet methylcytosine dioxygenase 2 (TET2) is a member of the methylcytosine dioxygenase enzyme family, which plays a role in the oxidative demethylation of 5-methylcytosine [6]. TET2 has been implicated in facilitating cell reprogramming by promoting the mesenchymal-to-epithelial transition (MET), a process opposite to EMT, wherein cells regain epithelial characteristics and apical-basal polarity. Hu et al. [7] found that TET2 promoted the MET-driven transformation of pluripotent stem cells via the miRNA200 family. However, the mechanism by which TET2 contributes to EMT in nasal epithelial cells remains unclear.
Numerous molecules have been identified that either directly or indirectly regulate the activity and expression levels of TET2. For instance, vitamin C (VC) has been shown to enhance the activity and expression of TET2, acting as an activator of TET enzymes. Conversely, hypoxia appears to downregulate TET2 expression [8]. Since TET2 is an oxygen- and α-ketoglutaratedependent enzyme that functions aerobically [9], it has been demonstrated to be regulated by both hypoxia and hypoxia-inducible factor 1α (HIF1α) [10]. Hypoxia, a factor known to influence glucose uptake, metabolism, angiogenesis, cell proliferation, differentiation, and apoptosis by activating the expression of related genes [11], has also been shown to impact the development of nasal polyps (NP). HIF1α, which increases in hypoxic environments, has been found to be highly expressed in the nasal epithelium of NP patients and can induce EMT in nasal epithelial cells, thereby influencing the pathogenesis of NP [4,12]. Additionally, vascular endothelial growth factor levels are elevated in NP-derived fibroblasts through HIF1α, contributing to the development of NP [13]. Hypoxia may also promote EMT in NP by inducing reactive oxygen species (ROS) and the secretion of high-mobility group box 1 (HMGB1) [14,15]. However, the relationship between EMT and CRSwNP, mediated by HIF1α and its downstream molecules, requires further investigation. Therefore, our study aimed to explore the role of TET2 in NP development through HIF1α-stimulated EMT.
MATERIALS AND METHODS
Patient tissue samples
Eleven NP tissue samples from CRSwNP patients, eight uncinate process from CRSsNP patients and 10 inferior turbinates from normal patients were collected from enrolled patients during endoscopic surgery from the Department of Otolaryngology, Renmin Hospital of Wuhan University (Wuhan, China) between January and December 2021. This research was approved by the Ethics Committee of Renmin Hospital (No. WDRY2021-K084) and the Animal Ethics Committee of Renmin Hospital (License No. WDRM 20211005). Before all experiments, informed consent was obtained from all the patients and their families.
The inferior turbinates of patients with nasal septal deviation or nasal fracture without sinus inflammation during nasal septoplasty and nasal bone reduction surgery were used as controls. The diagnosis of CRSwNP and CRSsNP were confirmed according to the diagnostic criteria from published guidelines [16]. Patients with any of the following criteria were excluded: (1) younger than 18 years old; (2) diagnosed with primary ciliary dyskinesia, posterior nostril polyps, fungal rhinosinusitis, cystic fibrosis, or a systemic coagulation disorder; (3) aspirin-sensitivity; (4) treated with antibiotics, glucocorticoids, or immune-modulating drugs for 4 weeks before surgery; and (5) immune system diseases and acute infection. The demographic and clinical information of all subjects were summarized in Table 1.
Immunohistochemical staining
The tissues from patients and mice were fixed with phosphate buffered saline (PBS) containing 4% formaldehyde for 24 hours (4 °C, pH 7.3), dehydrated in a graded ethanol series (70%, 80%, 90%, 95%, and 100%), embedded in paraffin, and then sliced into sections of 5-μm thickness and placed on poly-L-lysine–coated glass slides. The paraffin glass slides of CRSwNP, CRSsNP and control for immunohistochemical staining (IHC) were heated at 65 °C for 1 hour to dewax. Next, these slides were deparaffinized and rehydrated in a graded ethanol series (100%, 90%, 80%, and 70%), then incubated with 3% H2O2 for 10 minutes to block endogenous peroxidases, washed three times with PBS, the membrane was broken with 0.5% Triton X-100 (G1204, Servicebio), then subjected to heat-induced epitope retrieval and washed three times with PBS. Bovine serum albumin protein was applied to saturate any excess protein-binding sites for 30 minutes. The blocked sections were incubated with primary antibodies as following: E-cadherin (20874-1-AP, diluted 1:5,000; Proteintech), vimentin (10366-1-AP, diluted 1:5,000; Proteintech), TET2 (21207-1-AP, diluted 1:500; Proteintech), HIF1α (20960-1-AP, diluted 1:1,000; Proteintech), and Nrf2 (16396-1-AP, diluted 1:1,500; Proteintech) at 4 °C overnight. After the slides had reached the room temperature, washed these tissues with PBS for three times, a detection system of Streptomyces ovalbumin (OVA)-biotin method was used (SP-9001, ZSGB-BIO). Then the slides were visualized with diaminobenzidine color according to the manufacturer’s instructions. Next, the slides were stained with hematoxylin, dehydrated in a graded ethanol series, and xylene transparent. Finally, the slides were observed under a light microscope and integrated optical density (IOD) of different markers in nasal epithelium per area were further analyzed by Image-Pro Plus version 6.0 software (Media Cybernetics).
A murine NP model
Wild-type C57BL/6J mice (6 weeks old, male or female) were provided by Wuhan Shulaibao Biotech Co. Ltd. and kept in specific pathogen-free rooms. A murine NP model was generated from an allergic rhinosinusitis model in mice as described previously [4]. Mice were firstly immunized with an intraperitoneal injection of 25 mg of OVA (Sigma) in 2 mg of aluminum hydroxide gel on days 0 and 5, then treated with a daily intranasal instillation from days 12 to 19 with 6% OVA diluted in PBS. Next from days 20 to 47, mice were treated with 6% OVA 3 times a week to get continuous inflammation. At day 48, all groups of mice were challenged three times a week with 10 ng of Staphylococcus aureus enterotoxin B (SEB) plus 6% OVA for 8 weeks. The control groups were designated as follows: instillation with PBS only (negative control) and instillation with 6% OVA plus 10 ng of SEB (NP control). The experimental groups were designated as follow: instillation with 6% OVA plus SEB plus chemical (10 mg/kg VC) (see corresponding figure for detailed schedule). The sinonasal specimens were collected and processed by IHC, hematoxylin and Eosin (H&E) and periodic acid-Schiff (PAS) staining. Polyp formation and epithelial disruption markers were microscopically examined from coronal sections. The criteria for NPs included (1) more elevated lesion than surrounding mucosal folds, (2) eosinophilic infiltration, and (3) microcavities.
H&E and PAS staining
Nasal tissues from mice were fixed, embedded and made into paraffin sections. After rehydration, the tissue sections were stained with hematoxylin and eosin. The specific characteristics of goblet cells in mouse nasal mucosa were stained using PAS. The slides were observed under a light microscope. Number of polyps and count of red goblet cells were further analyzed by Image-Pro Plus version 6.0 software.
Enzyme-linked immunosorbent assay
The levels of tumor necrosis factor (TNF)-α, interleukin (IL)-6, and OVA-specific immunoglobulin E (IgE) of mice nasal lavage fluid were determined using enzyme-linked immunosorbent assay (ELISA) kits (Pyram) according to the manufacturer instructions. Briefly, the samples and biotin-conjugated antibodies were applied to ELISA microplates and then detected by using a microplate reader at 450 nm. The concentrations of target protein were calculated by a standard curve according to the manufacturer’s instructions.
Cell culture
Human nasal epithelial cells (hNECs) were scraped from the patient’s nasal polyps, and sent back to lab in airway epithelial cell growth medium. After cells were centrifuged at 1,000 rpm for 5 minutes, the supernatant was removed, the cells were resuspended and then cultured in Bronchial Epithelial Cell Growth Medium BulletKit from Lonza (CC-3170). hNECs were used for analysis within five passages.
Cell counting kit-8 assay
hNECs were suspended and seeded into 96-well plates. After being incubated at 37 °C with 50 μM, 150 μM, 250 μM, and 500 μM CoCl2 (Aladdin, C106772) for 72 hours, 10 µL of cell counting kit-8 (CCK-8) solution (G4103, Servicebio) was added to each well. The absorbance was measured by a microplate reader at 450 nm after being incubated for 2 hours in a dark environment.
Implementation of CoCl2-induced hypoxia
EMT of hNECs was induced by treatment with 150 μM CoCl2 (concentration determined by CCK-8) for 72 hours. Then, VC (Aladdin, A103539) was also added into CoCl2-stimulated hNECs for 72 hours. During this period, medium needs to be refreshed every day until the time is up to 72 hours. CoCl2 with or without VC were added soon after medium changed.
Small interfering RNA transfection
Cells were transfected at time of seeding with 10 nM non-targeting control small interfering RNA (siRNA) and 20 nM siTET2 (GenePharma) using Lipofectamine RNAimax in Bronchial Epithelial Cell Growth Basal Medium media following manufactures protocol. 1.5×105 cells were seeding into 35 mm plates with a fresh media change after attachment. After 48 hours transfection, cells were then stimulated with 150 μM CoCl2 (C106772, Aladdin) and 100 μM VC (A103539, Aladdin) for 72 hours and collected for further analysis.
Western blotting
Control tissues, CRSwNP tissues, CRSsNP tissues, and incubated cells were dissected and lysed in radioimmunoprecipitation assay lysis solution (Beyotime) containing phosphatase inhibitors and PMSF for at least 15 minutes. The protein concentrations were measured using the bicinchoninic acid protein assay kit (G2026, Servicebio). Thirty-microgram protein was loaded on 10% sodium dodecylsulfate–polyacrylamide gel. The proteins in the gel were then transferred to polyvinylidene difluoride membranes for 2.5 hours and were incubated in blocking solution (5% non-fat milk in Tris Buffered Saline with Tween) for 1 hour at room temperature. Then the proteins were incubated overnight at 4 °C with primary antibodies, including E-cadherin (20874-1-AP, Proteintech), vimentin (10366-1-AP, Proteintech) in a dilution ratio of 1:5,000, and HIF1α (20960-1-AP, Proteintech), TET2 (21207-1-AP, Proteintech), Nrf2 (16396-1-AP, Proteintech) in a dilution ratio of 1:1,500, α-SMA (14395-1-AP, Proteintech) and β-actin (GB15003, Servicebio) in a dilution ratio of 1:1,000. The membranes were washed three times with TBST and incubated with secondary antibodies, horseradish peroxidase-conjugated goat anti-rabbit IgG (SA00001-2, Proteintech, diluted 1:3,000) for 60 minutes at room temperature. An electrochemiluminescence kit (BL523B, Biosharp) was used to visualize the membranes. The relative expression level of proteins was represented in the ratio of the target protein and β-actin using Fiji software (National Institutes of Health).
Immunofluorescence
For immunofluorescence cytochemistry, hNECs were incubated with or without CoCl2 or VC in six-well plates for 72 hours. Next, the cells were fixed with 4% paraformaldehyde at room temperature for 15 minutes, washed with PBS three times. Next, the cells were incubated with blocking solution (10% goat serum with PBS) for 30 minutes at room temperature, then incubated with primary antibody (E-cadherin, vimentin) overnight at 4 °C. After rinsing, the sections and cells were incubated with Goat anti-Rabbit IgG-Alexa Fluor 488 diluted 1:200 (abs20025, Absin) for 1 hour, and counterstained with SuperKine Enhanced Antifade Mounting Medium with DAPI (Abbkine) for the staining of nuclei. Images were taken with a fluorescence microscopy. Relative fluorescence expression of E-cadherin and vimentin (Intensity/Area) were further analyzed by Fiji software.
Assessment of ROS
Intracellular ROS level was determined using DCFH-DA (S0033S, Beyotime Institute of Biotechnology) according to the manufacturer’s instructions.
Bisulfite sequencing polymerase chain reaction
DNA methylation was examined by bisulfite conversion of extracted genomic DNA with a bisulfite conversion kit (DP215, TIANGEN). The Nrf2 promoter was amplified by polymerase chain reaction (PCR) using a methylation-specific PCR kit (EM101, TIANGEN). Primer sequences were designed using Methyl Primer Express v1.0 (Applied Biosystems), and Sanger sequencing was performed on the PCR products. Primers were designed as follows: Nrf2-bisulfite sequencing PCR (BSP) forward: 5´-GTTTGTTGGGATTTTAGTTGGTAGTT-3´; Nrf2-BSP reverse: 5´-CCAAATCCATCATAATAAACTATAAACC-3´.
Statistical analysis
All data were analyzed with GraphPad Prism (ver. 9.1). The measured data were presented as the mean±standard deviation. Quantitative data were analyzed using the unpaired Student t-test for comparing two groups and one-way analysis of variance for multiple groups if the data follows a normal distribution. Categorical data were analyzed using chi-square test. Pearson correlations were used to determine variable relationships. Significance was considered when the P-value was less than 0.05.
RESULTS
TET2 and HIF1α expression in nasal tissue epithelium
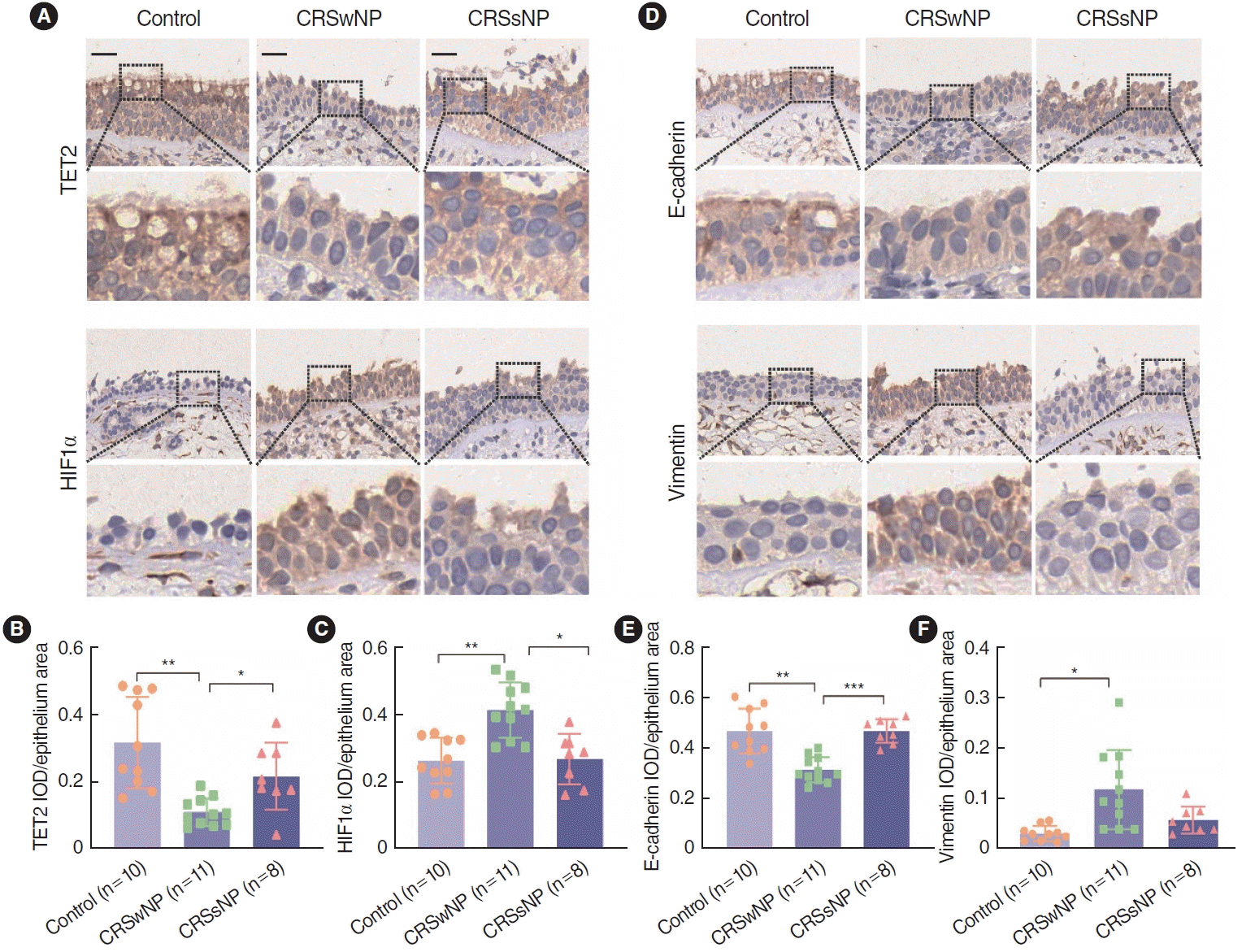
To evaluate the expression of TET2 and HIF1α in nasal epithelial tissue, we performed IHC staining on 10 control tissue samples, 11 CRSwNP tissue samples, and 8 CRSsNP tissue samples, focusing on the expression patterns of these proteins in the nasal epithelium (Fig. 1A). Using a semiquantitative evaluation of the IOD in the epithelial area, we found that HIF1α expression was significantly upregulated in the CRSwNP tissues, while TET2 expression was significantly downregulated (Fig. 1B and C). Additionally, IHC staining for E-cadherin and vimentin is presented in Fig. 1D. E-cadherin expression was significantly reduced in the CRSwNP group compared to both the control and CRSsNP groups, whereas the expression of the mesenchymal marker vimentin was increased in the CRSwNP group (Fig. 1E and F). Collectively, these results indicate that HIF1α levels were elevated in NP nasal epithelium, but TET2 expression was significantly diminished. Therefore, it is speculated that the expression of TET2 and HIF1α in the nasal epithelium may play an important role in the pathogenesis of CRSwNP.
Correlations among EMT markers, TET2, and HIF1α in the nasal epithelium
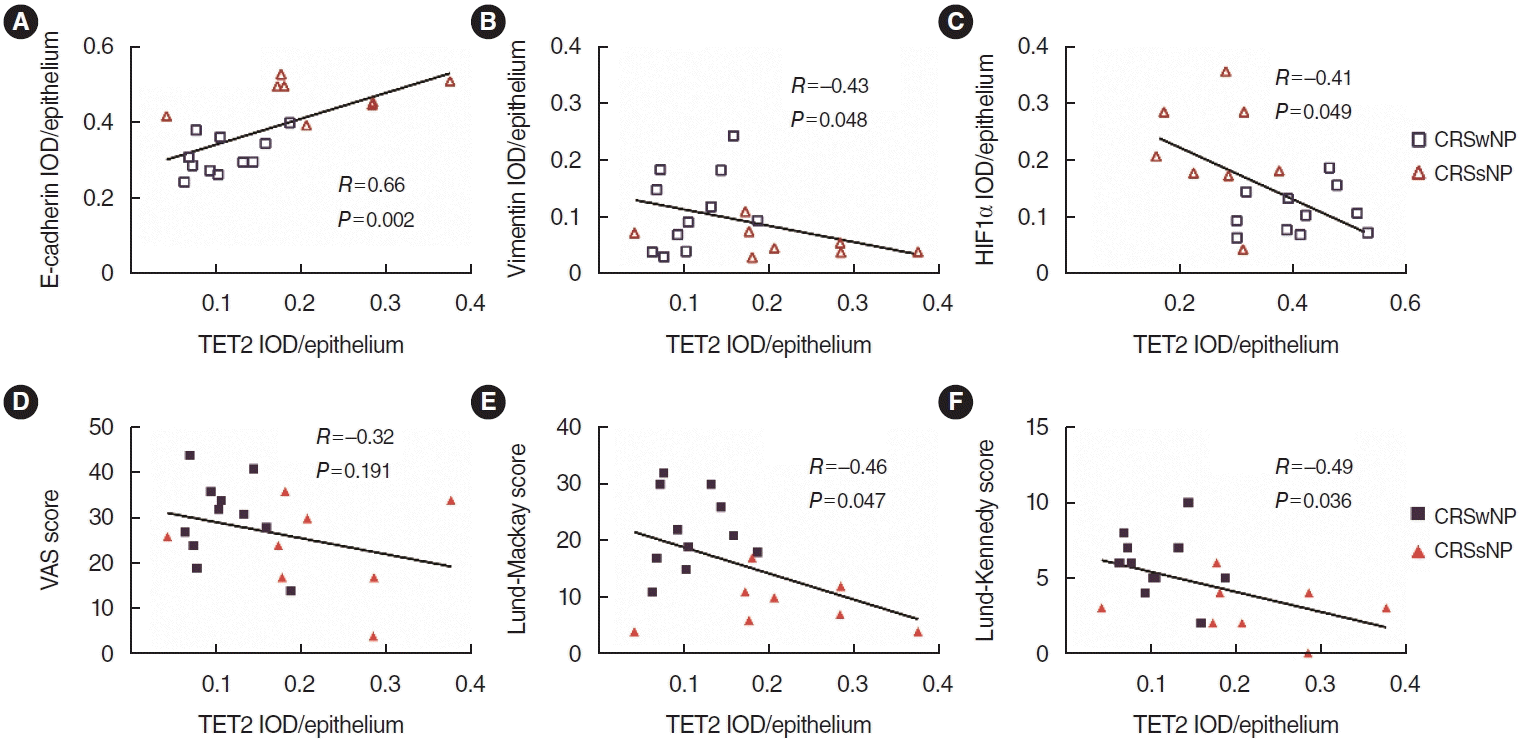
We also investigated correlations among EMT markers, TET2, and HIF1α in the nasal epithelium. The results of the correlation analysis suggested that TET2 expression levels in the nasal epithelium were positively correlated with E-cadherin (Fig. 2A) and negatively correlated with vimentin (Fig. 2B). Furthermore, we analyzed correlations between TET2 expression in the nasal epithelium and clinical scales relevant to disease severity parameters, such as the visual analog scale (VAS) score, the Lund-Mackay score, and the Lund–Kennedy score. TET2 expression levels were negatively correlated with both the Lund-Mackay score (Fig. 2E) and the Lund-Kennedy score (Fig. 2F). However, there was no significant relationship between TET2 and the VAS score (Fig. 2D). Interestingly, we found that TET2 expression was negatively correlated with HIF1α expression in the nasal epithelium (Fig. 2C). Taken together, these results suggest that TET2 expression is related to the development of CRSwNP and that there may be an association between TET2 and HIF1α in nasal polyps, potentially by inducing or inhibiting the EMT.
TET2 and HIF1α expressions in a murine model
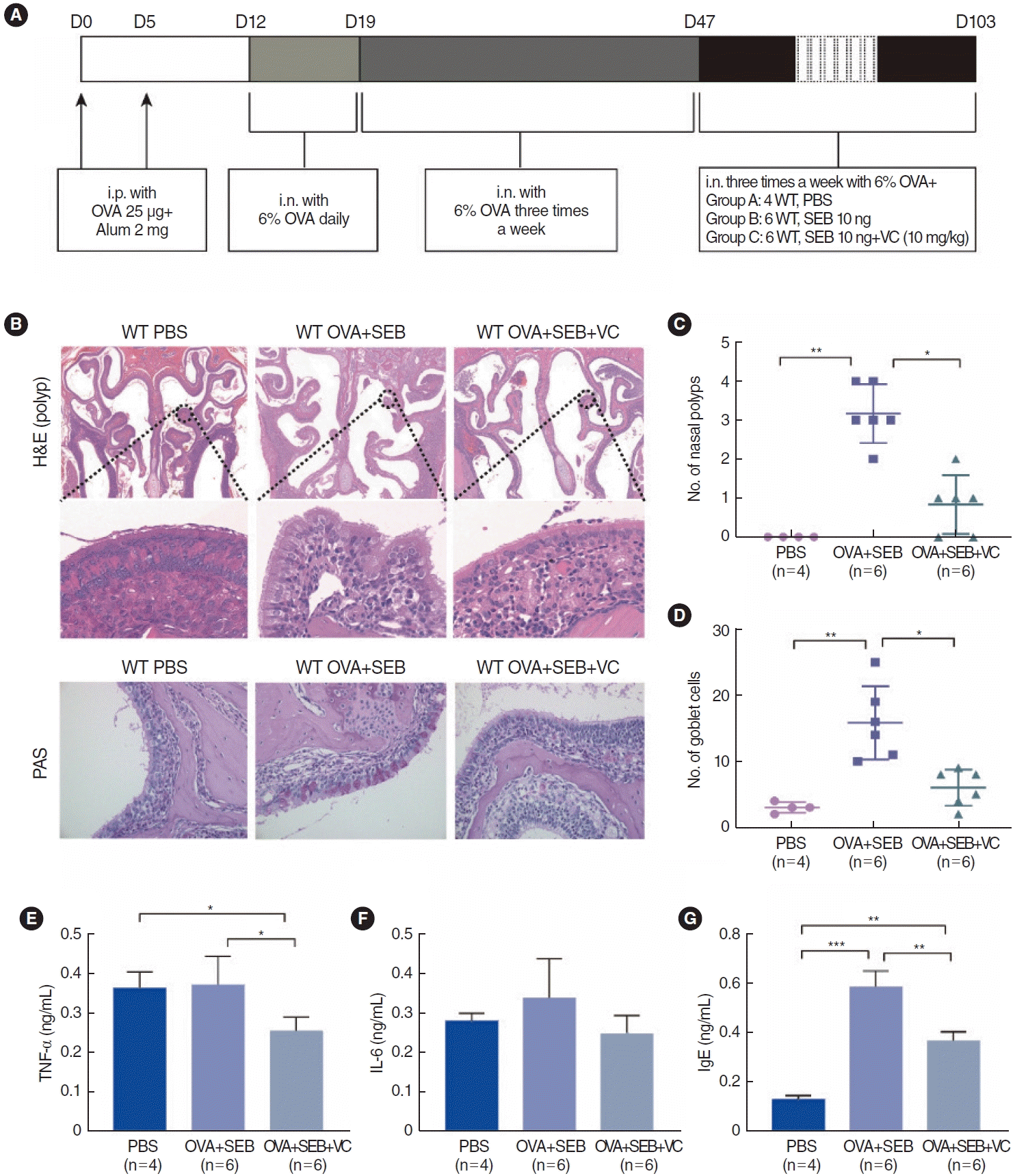
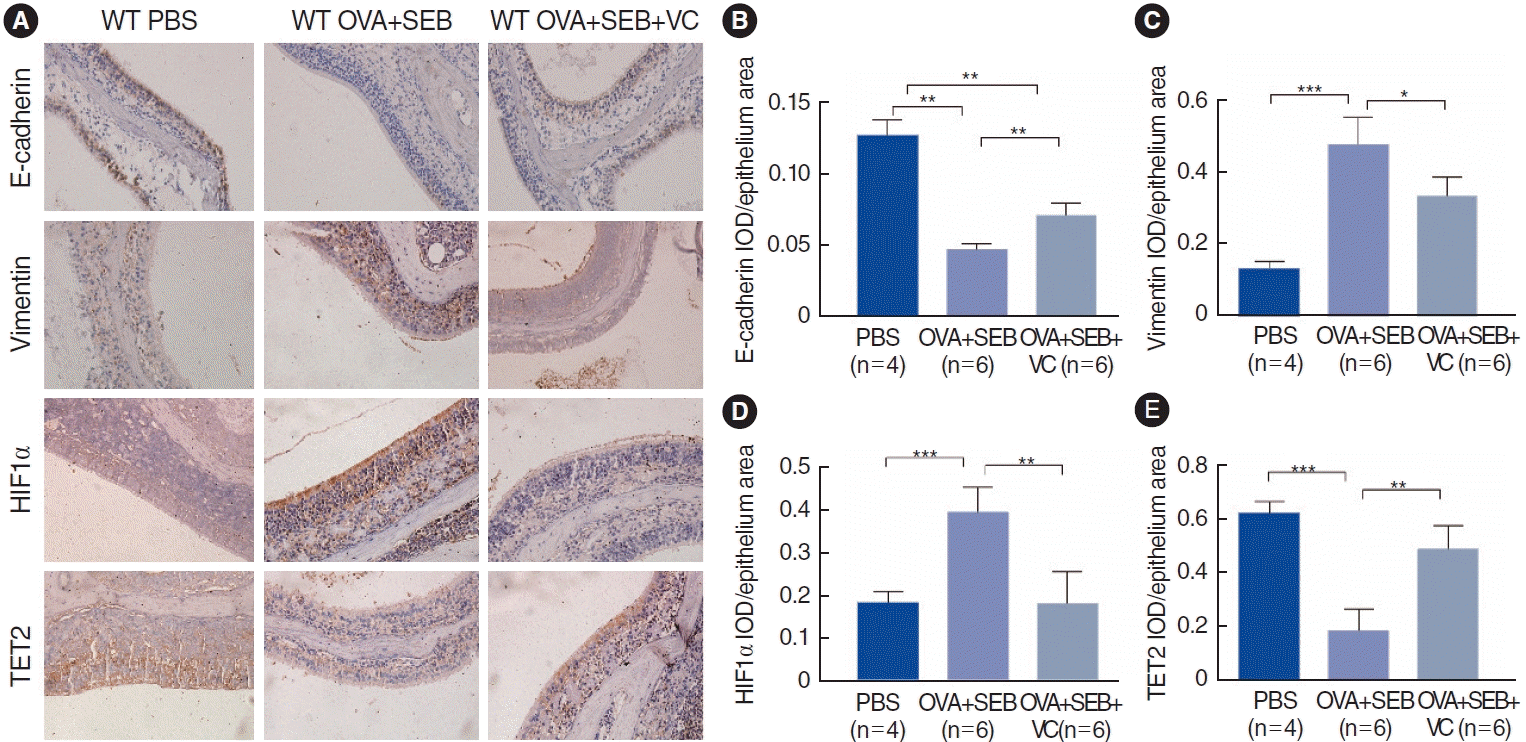
To further investigate TET2 and HIF1α expression in vivo, a murine model of NP was established using wild-type mice according to an established reported protocol (Fig. 3A). OVA and SEB treatment led to prominent polypoid lesions with increased infiltration of inflammatory cells and an increase in goblet cells when compared with the PBS group (Fig. 3B-D). In the OVA+SEB group, OVA-specific IgE levels were significantly elevated in nasal lavage fluid, and the inflammatory cytokine TNF-α was also upregulated. However, IL-6 showed a non-significant difference when compared with the PBS group. TET2 activation by VC significantly decreased the levels of OVA-specific IgE and TNF-α (Fig. 3E-G). Additionally, epithelial disruptions, indicated by reduced E-cadherin, and TET2 expression were significantly more pronounced in the OVA+SEB group than in the PBS group, while HIF1α expression and vimentin levels were increased (Fig. 4A-E). Treatment with the TET2 activator VC resulted in a significant increase in E-cadherin and TET2 expression, whereas HIF1α and vimentin expression were decreased (Fig. 4A-E). Taken together, these findings suggest that TET2 activation by VC may reduce nasal polyp formation and nasal inflammation concurrently in vivo.
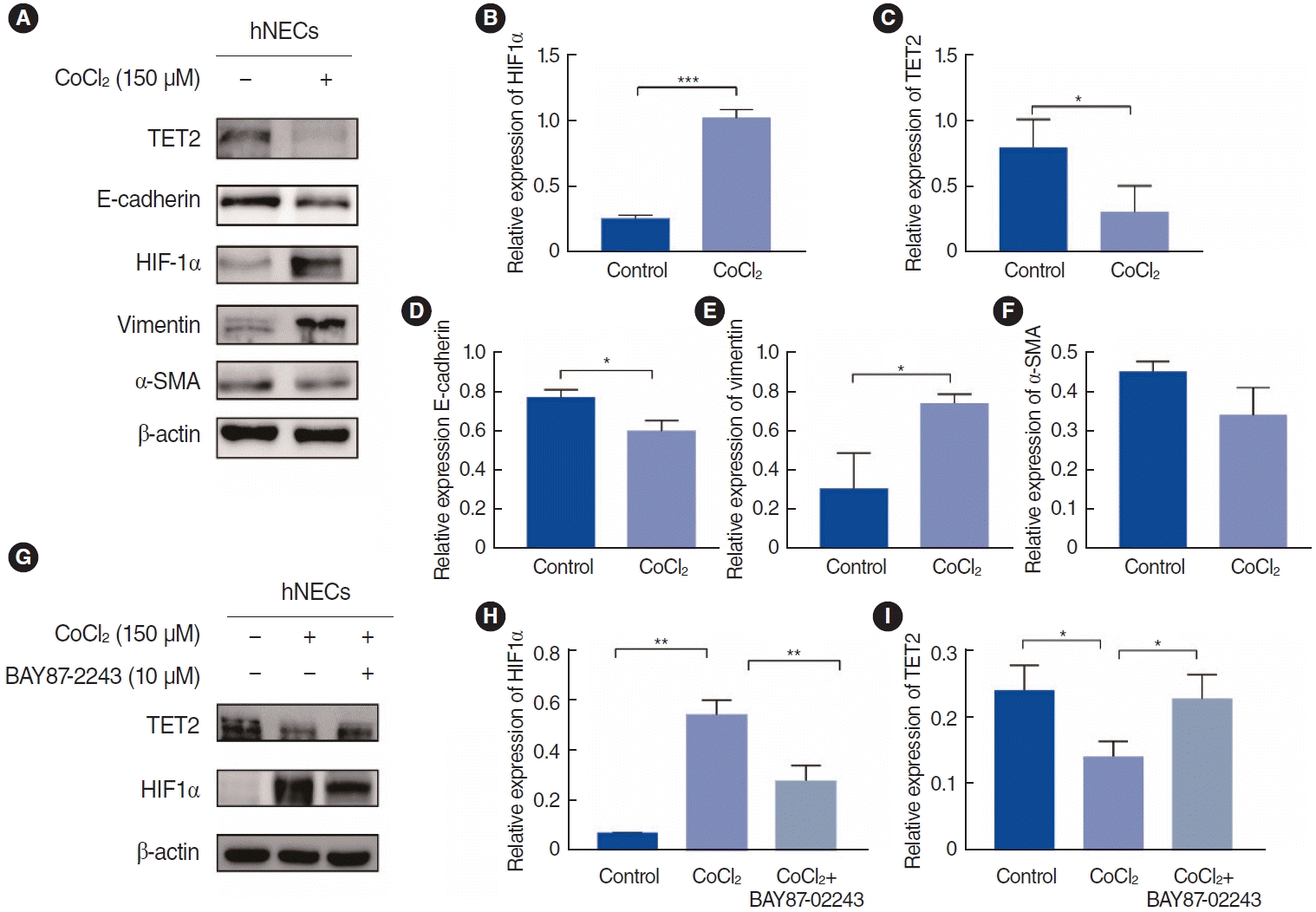
TET2 was regulated by HIF1α in an ex vivo EMT model in hNECs via CoCl2-stimulated hypoxia
In our study, CoCl2 was used to mimic hypoxia in hNECs at a concentration of 150 µM, as determined by a CCK-8 cell viability assay (Supplementary Fig. 1), for a duration of 72 hours [12]. Subsequently, we assessed the expression of EMT markers, TET2, and HIF1α by Western blotting (Fig. 5A). We observed that CoCl2- induced hypoxia significantly reduced the levels of the epithelial marker E-cadherin and TET2 (Fig. 5B and C). Conversely, the mesenchymal marker vimentin and HIF1α were upregulated (Fig. 5D-F), but not α-SMA. Previous research has indicated that α-SMA levels increase significantly in hypoxic hNECs compared to controls [4]. However, in our study, α-SMA did not exhibit significant changes, which may be attributed to a relatively high baseline level of α-SMA that precluded the detection of statistically significant alterations. To elucidate the relationship between HIF1α and TET2, we employed the HIF1α-specific inhibitor BAY87-2243 (Fig. 5G and H). Our findings revealed an increase in TET2 expression after 72 hours of hypoxia (Fig. 5I), suggesting that TET2 is regulated by HIF1α. This relationship between TET2 and HIF1α was further substantiated in hypoxic hNECs, confirming that HIF1α modulates TET2 expression.
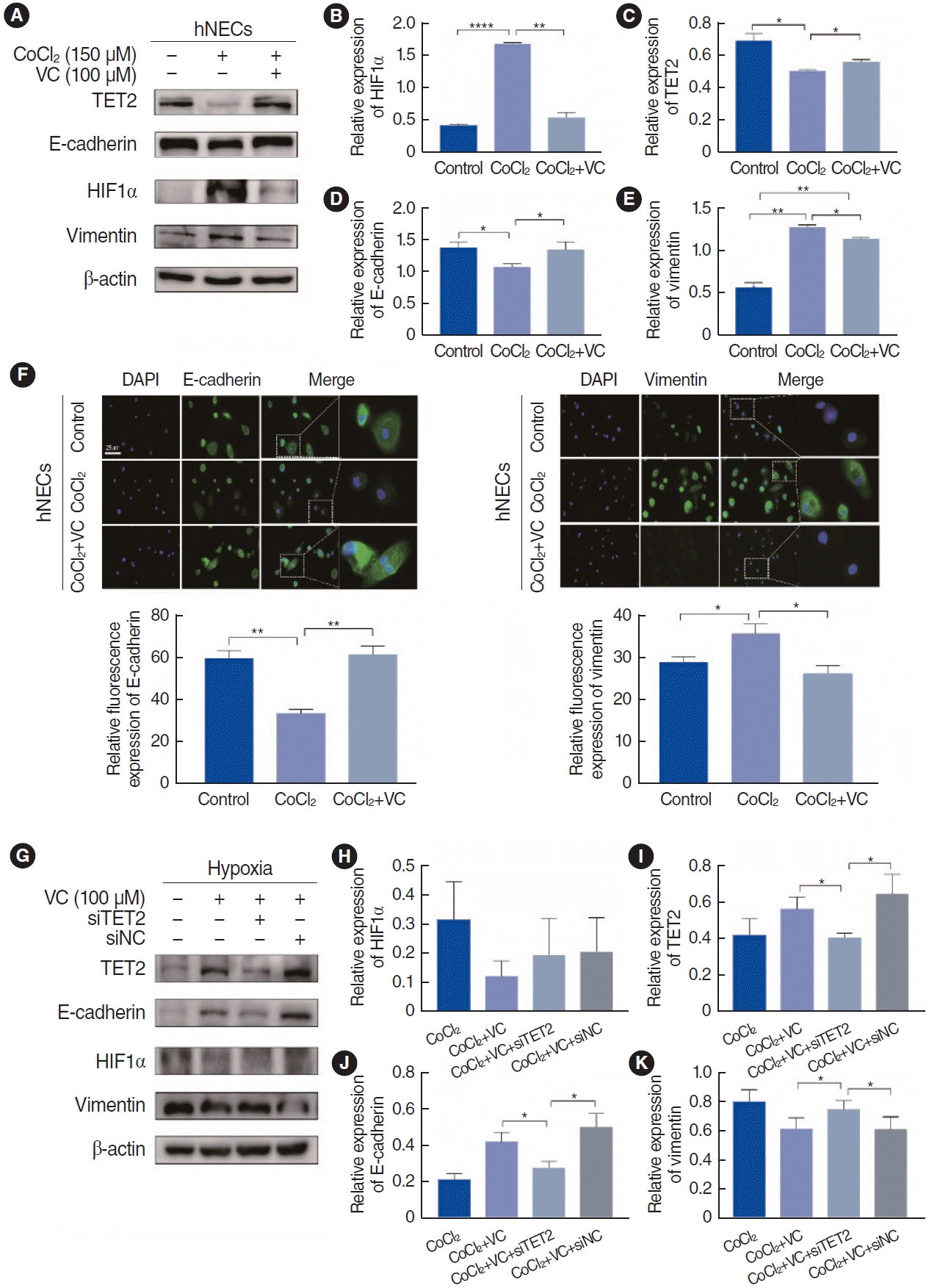
Activation of TET2 inhibited EMT in CoCl2-treated hypoxic hNECs
As previously mentioned, VC acts as a specific activator of TET2 and can initiate the reversal of EMT at a concentration of 100 µM in breast cancer cells [17]. We used this same concentration to investigate the activation of TET2 by VC in CoCl2-treated hNECs. VC successfully activated TET2 expression and significantly downregulated HIF1α in hypoxic hNECs (Fig. 6B and C). Additionally, treatment with 100 µM VC and 150 µM CoCl2 for 72 hours increased the expression of E-cadherin in hNECs, while a reduction in vimentin expression was observed in both immunofluorescence examination and Western blotting (Fig. 6A and D-F). Given that VC is a nonspecific activator, we employed siRNA technology to silence TET2 expression. As depicted in Fig. 6I, the ability of VC to reverse EMT in CoCl2-treated hNECs was lost (Fig. 6J and K) when TET2 was knocked down (Fig. 6G), suggesting that VC inhibits EMT by activating TET2. However, the level of HIF1α showed no significant difference (Fig. 6H), further explaining that HIF1α might be the upstream of TET2. In summary, TET2 can inhibit EMT in CoCl2-treated hypoxic hNECs.
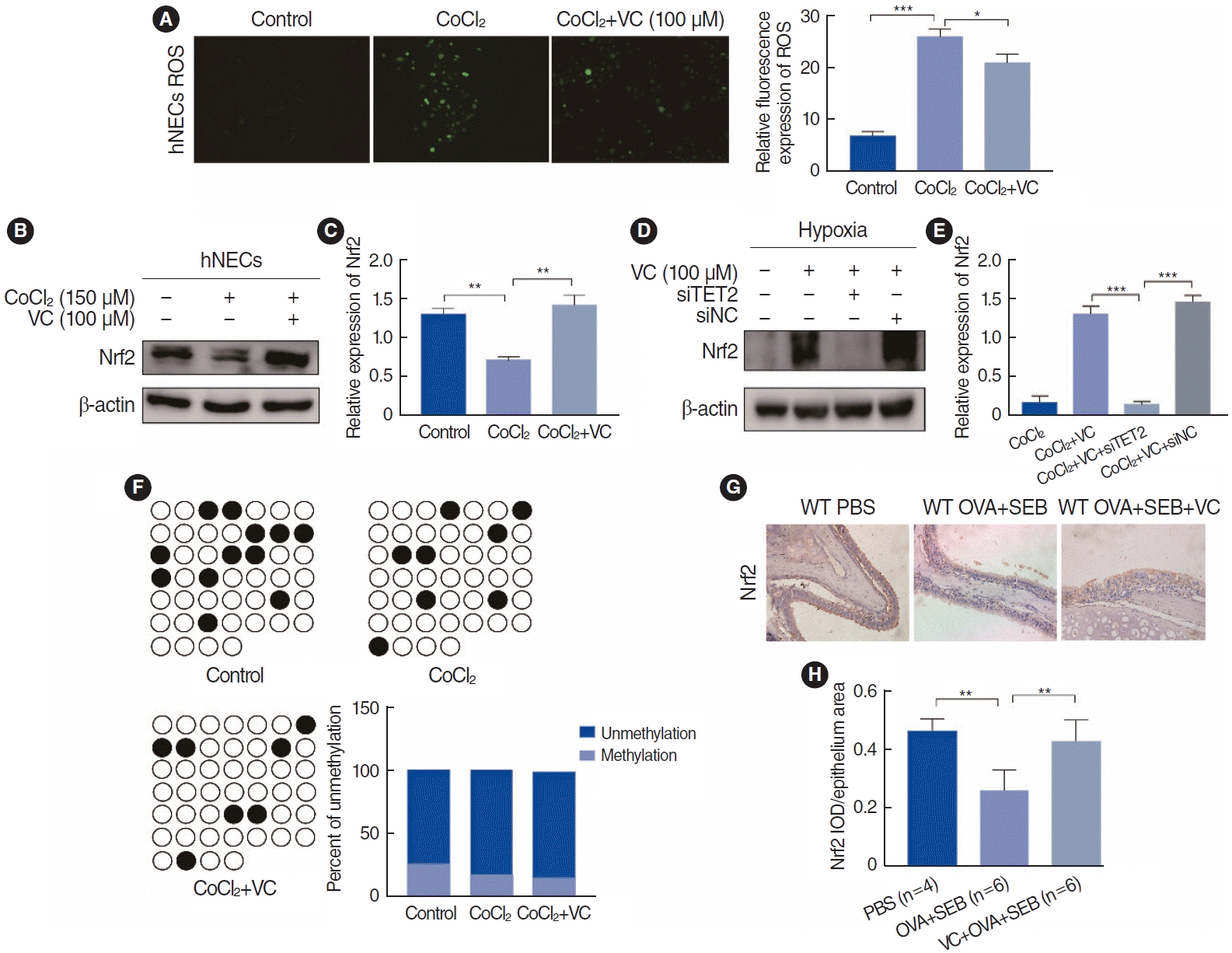
TET2 induced Nrf2 expression to inhibit HIF1α-mediated EMT in hNECs
Hypoxia may also contribute to nasal polyp EMT by inducing ROS [14,15]. Nrf2, an inhibitor of ROS, plays a role in regulating the EMT by modulating the ROS-mediated PI3K/Akt/GSK-3β pathway in renal tubule cells [18]. In our study, we demonstrated that ROS generation was elevated in CoCl2-induced hypoxic hNECs, but TET2 activation reduced ROS levels (Fig. 7A). Concurrently, Nrf2 expression was significantly lower in CoCl2- treated hypoxic hNECs, while TET2 activation markedly increased Nrf2 expression (Fig. 7B and C). However, after TET2 silencing, VC lost its target and could not increase Nrf2 expression through its antioxidant effect (Fig. 7D and E), suggesting that Nrf2 is a downstream target of TET2. We also analyzed three samples to assess the methylation status of the Nrf2 promoter, but there were no significant differences in the number of CpGs among the groups (Fig. 7F). Additionally, we examined Nrf2 expression in a murine nasal polyp model (Fig. 7G) and found that Nrf2 was significantly upregulated in vivo when TET2 was activated, compared with the OVA+SEB group (Fig. 7H). These findings suggest that TET2 may inhibit HIF1α-mediated EMT in hNECs by inducing Nrf2 expression.
DISCUSSION
The EMT is a cellular program in which epithelial cells transiently transition into a mesenchymal cell state [19]. Driven by specific regulators, the EMT contributes to the remodeling of upper airway tissues [20,21], a process that has also been implicated in the tissue remodeling associated with CRSwNP [22].
Hypoxia and the stable expression of HIF1α have been implicated in the processes of the EMT, immune response, and inflammatory pathways in CRSwNP [23]. Immunohistochemical and Western blot analyses in our study revealed increased HIF1α expression in patients with CRSwNP. Additionally, correlation analysis confirmed that HIF1α expression was positively associated with vimentin and inversely associated with E-cadherin. In vitro studies have shown that nasal epithelial cells can undergo EMT when exposed to hypoxic conditions for 72 hours [4,12]. In this study, we used CoCl2 to simulate chemical hypoxia and induce the EMT in nasal epithelial cells. Our findings indicate that hNECs can transition to a mesenchymal state after 72 hours of hypoxia. Collectively, these results suggest that HIF1α contributes to the development of NP through EMT. However, the downstream signaling molecules through which HIF1α regulates the EMT in CRSwNP and nasal epithelial cells remain unclear.
Emerging evidence suggests that epigenetic regulation plays a role in the EMT process [24]. TET enzymes, which are epigenetic modifiers, have been shown to influence various types of tumorigenesis, including endometrial cancer [25], ovarian cancer [26], and breast cancer [27], by inhibiting EMT. Similarly, genetic and epigenetic factors have been reported to determine the EMT and tissue remodeling in NP [28]. In our study, we observed that TET2 expression was lower in the nasal epithelium of patients with CRSwNP than in controls and the CRSsNP groups. Furthermore, TET2 expression was positively correlated with E-cadherin expression and negatively correlated with vimentin expression, indicating a strong relationship between TET2 and the EMT. Additionally, our correlation analysis suggested that TET2 expression in the nasal epithelium was inversely related to HIF1α. We also discovered that in a mouse model of NP, HIF1α was significantly upregulated, while TET2 was significantly downregulated in the nasal epithelium. The precise nature of the relationship between TET2 and HIF1α in the regulation of EMT in NP epithelium requires further investigation.
CRSwNP is a disease characterized by chronic inflammation of the nasal mucosa, involving a variety of inflammatory cells. Dysfunction of the nasal epithelium barrier, triggered by factors such as allergens, bacteria, and hypoxia, not only contributes to the EMT but also to local chronic inflammation. Several studies have identified TET2 as playing a crucial role in both the initiation and resolution of inflammation. During pathogen infection, TET2 can directly reduce the 5-methylcytosine level of Socs3, a negative regulator of the JAK-STAT pathway, thereby activating the emergency production of mature innate immune cells [29]. TET2 has also been shown to bind and recruit HDAC1/2, facilitating histone deacetylation and suppressing IL-6 expression during the resolution phase of inflammation in innate myeloid cells [30]. In the context of airway inflammation, TET2 has been reported to modulate lipid peroxidation and ferroptosis in airway epithelial cells, thereby alleviating inflammation caused by cigarette smoke in chronic obstructive pulmonary disease mice [31]. Our previous studies have demonstrated that a loss of TET2 results in exacerbated allergic inflammation and an imbalance between Th1/Th2 and Treg/Th17 cells in patients with allergic rhinitis [32,33]. Although TET2 appears to be implicated in the pathogenesis of upper airway inflammation, its precise role in the inflammation initiation and resolution processes of CRSwNP remains to be elucidated.
Since TET2 is an oxygen-dependent enzyme, numerous studies have focused on the relationship between TET enzymes, hypoxia, and HIF1α. In tumors, hypoxia can lead to DNA hypermethylation by reducing TET activity [27]. It has been found that TET2 antagonizes the function of HIF1/2α in metabolic reprogramming, thereby inhibiting the growth of clear cell renal cell carcinoma [34]. This suggests that HIF1α may act as a downstream molecule of TET2. However, other studies have shown that silencing HIF1α can restore TET2 expression in hypoxic human metastatic melanoma cells [35], which seems to indicate that HIF1α could be upstream of TET2. To date, the regulatory mechanism between HIF1α and TET2 expression is not fully understood. Our in vitro experiments suggested that TET2 expression in hNECs was downregulated by CoCl2-induced hypoxia. Subsequently, we applied a HIF1α-specific inhibitor and found that TET2 expression increased after 72 hours of hypoxia, implying that TET2 is regulated by HIF1α. In summary, hypoxia induces HIF1α expression, and HIF1α simultaneously decreases TET2 expression, thus contributing to EMT in nasal epithelial cells.
TET2-mediated demethylation plays a role in the protective effect of triptolide on podocytes by inhibiting the EMT [36]. Furthermore, TET2 is essential for controlling the growth and metastasis of melanoma in mice, as it directly counteracts TGF-β1-induced EMT [37]. In our study, we discovered that VC, a non-specific activator of TET2, restored TET2 expression, leading to the reversal of the EMT and a reduction in HIF1α levels in both hNECs and NP mice. In cancer research, high concentrations of VC alone can inhibit the invasion and metastasis of pancreatic ductal adenocarcinoma cells by blocking Wnt/β-catenin-mediated EMT [38]. To rule out the direct effects of VC and its non-specific action on TET2, we knocked down TET2 expression and observed that VC’s ability to reverse the EMT was significantly diminished in CoCl2-induced hNECs following TET2 gene silencing. This suggests that VC influences the EMT by activating TET2 in hypoxic hNECs. However, the exact mechanism by which TET2 regulates EMT in hNECs under hypoxic conditions remains to be clarified.
Oxidative stress has been implicated in nasal polypogenesis. Elevated levels of oxidative stress markers, such as thioredoxin-interacting proteins (TXNIP) and malondialdehyde, have been observed in patients with CRSwNP, while levels of the antioxidative stress molecule superoxide dismutase were found to be decreased in these patients [39]. Compared to the control group, nicotinamide adenine dinucleotide phosphate—which promotes the production of ROS—and the oxidative stress marker 4-hydroxy-2-nonenal were highly expressed in CRSwNP tissues [40]. These findings collectively underscore the significant role that oxidative stress plays in the pathogenesis of nasal polyps.
Nrf2 is a well-documented stress-related transcription factor, with oxidative stress being one of its most relevant triggers. Numerous studies have demonstrated that hypoxia and HIF1α lead to oxidative stress, resulting in an increase in ROS. To counteract this harmful process, Nrf2 expression is also upregulated. However, is this the case in the nasal epithelium of CRSwNP patients? To date, there is limited evidence regarding the role of Nrf2 in CRS. Studies using epithelial-specific Nrf2 knockout mice have shown that these mice exhibit goblet cell proliferation and significant eosinophilic infiltration in the nasal epithelium following a papain-induced model of rhinosinusitis, compared to wild-type mice [41]. Additionally, Nrf2 activation has been shown to recover cigarette smoke-induced epithelial barrier dysfunction in the nasal mucosa [42]. These findings suggest that Nrf2 plays a role in mitigating inflammation and inflammatory cell infiltration in CRS. In our research, we investigated the expression of Nrf2 in hypoxic hNECs and in a mouse model of NP. We observed that Nrf2 expression was lower than that of the control in both in vivo and in vitro settings, while ROS levels were elevated in hNECs. Interestingly, the activation of TET2 by the addition of VC increased Nrf2 expression in both in vivo and in vitro experiments and concurrently decreased ROS generation in vitro. Furthermore, we discovered that VC lost its ability to restore Nrf2 expression when TET2 was silenced, indicating that Nrf2 acts as a downstream molecule of TET2. However, BSP-sequencing revealed no significant change in the number of CpGs in the Nrf2 promoter region after VC was added to CoCl2-induced hNECs, suggesting that TET2 might induce Nrf2 expression indirectly. Although we measured the methylation level at the Nrf2 promoter region, the hydroxymethylation level should also be evaluated in future studies. In conclusion, we identified that TET2 inhibits the EMT by inducing Nrf2 expression in CoCl2-simulated hypoxic hNECs.
Murine NP models induced by hypoxia are relatively scarce; therefore, we established a rhinosinusitis model in mice as previously described [4]. We employed a C57BL/6J murine NP model induced by OVA and SEB to investigate the roles of TET2 and EMT in our study, which likely represents an eosinophilic NP model. Although a neutrophilic murine NP model might have been more appropriate, our prior research indicated that the expression of EMT showed no significant difference between eosinophilic CRSwNP and non-eosinophilic CRSwNP groups in human subjects [43,44]. Therefore, we chose to develop this eosinophilic NP model to mirror the characteristics of this inflammatory disease. Recently, an increasing number of researchers have employed BALB/c mice sensitized with LPS to create CRSwNP animal models. Wee et al. [45] compared the histopathological profiles of epithelial disruptions and found no significant difference between the LPS+SEB group and the SEB group; however, they did not evaluate EMT markers. Moreover, C57BL/6J mice have also demonstrated similar polypoid changes in the sinonasal mucosa and inflammatory patterns to those observed in BALB/c mice [46]. In light of these findings, we chose to use the C57BL/6J murine NP model. Nonetheless, the relationship between C57BL/6J and BALB/c mice in representing different NP models requires further experimentation to elucidate the mechanisms underlying CRSwNP.
Our study has some limitations. First, the sample size for IHC staining was relatively small. Second, it would be beneficial to design a series of varying concentrations of VC to determine whether the activation of TET2 by VC in nasal epithelial cells is dose-dependent and to identify the most appropriate concentration. Third, our previous studies did not find a significant difference in the expression of EMT markers and TET2 between patients with eosinophilic CRSwNP and those with non-eosinophilic CRSwNP [44]; therefore, we did not categorize the patients into these two groups. However, further research is needed to ascertain whether there is a difference in TET2 expression between patients with eosinophilic CRSwNP and those with neutrophilic CRSwNP, which would deepen our understanding of the condition.
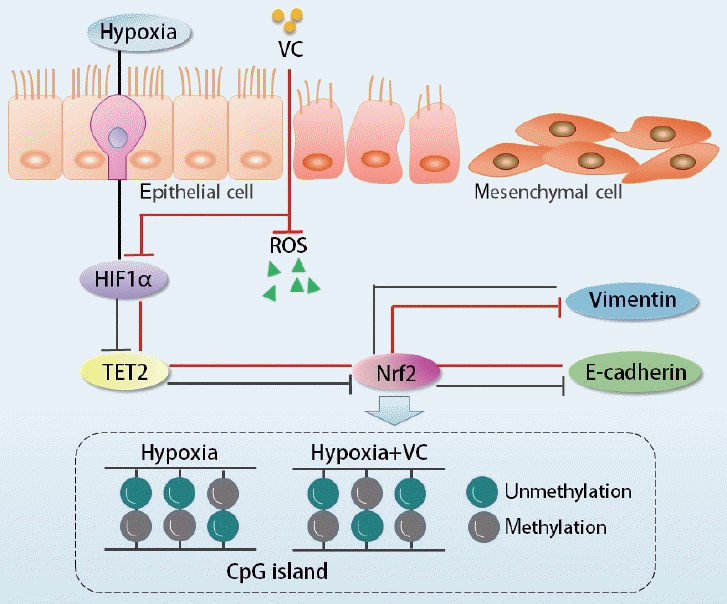
Taken together, our study demonstrated that TET2 expression was downregulated by HIF1α, which in turn regulated Nrf2 expression, ultimately leading to the EMT in CoCl2-induced hypoxic hNECs (Fig. 8). These findings indicate that the mechanisms involving TET2, Nrf2, and oxidative stress in the hypoxia-induced EMT of nasal epithelial cells warrant further investigation. More importantly, TET2 and its downstream molecules may represent therapeutic targets, offering a novel treatment strategy for nasal polyposis.
HIGHLIGHTS
▪ Tet methylcytosine dioxygenase 2 (TET2) expression was negatively correlated with epithelial-to-mesenchymal transition (EMT) markers and hypoxia-inducible factor 1α (HIF1α) expression in human nasal epithelium.
▪ The activation of TET2 by vitamin C alleviated nasal polyp formation and EMT in wild-type mice.
▪ In vitro experiments demonstrated that TET2 deficiency contributed to HIF1α-mediated EMT progress and aggravated the development of chronic rhinosinusitis with nasal polyps by partially facilitating the production of reactive oxygen species in human primary nasal epithelial cells.
 XML Download
XML Download