

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Head and neck squamous cell carcinoma (HNSCC) ranks among the most common cancers worldwide, with over 600,000 new cases reported each year [1]. The standard treatment for locally advanced HNSCC involves a combination of radiation therapy and cisplatin. However, despite aggressive multimodal therapy, the 5-year survival rate for these patients hovers around 50% [2]. Predictive and prognostic models play a critical role in the management and outcome prediction of cancer. The persistent lack of improvement in patient survival and the need for personalized treatment strategies have spurred research into the molecular underpinnings of HNSCC. It is therefore essential to develop a method that captures the global characteristics of HNSCC, is resilient to its heterogeneity, and yields clinically relevant insights. Classic prognostic indicators for HNSCC include a patient’s smoking history and human papillomavirus (HPV) status [3,4]. Beyond HPV status, however, no other biomarkers for HNSCC have been definitively established. Therefore, further research is needed to identify new prognostic markers that can better guide the stratification of treatment and prognosis for patients.
CXCL8 is a key biomarker closely associated with tumor development and progression. It has been consistently linked to the advancement of various cancers and significantly affects tumor growth and migration. In colorectal cancer, CXCL8 is vital within the tumor microenvironment (TME) and influences the response to immunotherapy, impacting immune evasion. It may also improve the prognosis of malignant glioma and contribute to the effectiveness of chemotherapy [5]. Elevated levels of CXCL8, along with CXCL10 and CXCL11, in breast cancer patients have been shown to correlate with reduced overall survival (OS), emphasizing its potential as both a therapeutic target and a prognostic marker for breast tumors [6]. In gastric cancer, increased CXCL8 expression is associated with lower relapse-free survival, underscoring its importance in disease progression and prognosis [7]. In recurrent ovarian cancer, CXCL8 expression is increased, whereas it appears to decrease in late-stage tumors, suggesting its utility as a diagnostic biomarker [8]. Additionally, in esophageal cancer patients, higher CXCL8 levels are positively correlated with the depth of tumor invasion and C-reactive protein (CRP) concentrations, further supporting its potential as a tumor marker [9].
CXCL8 also plays a significant role in tumor therapy. Notably, the CXCL8-CXCR1/2 axis is crucial for enhancing the efficacy of targeted therapies and chemotherapy [1]. The combined targeting of the CXCL8-CXCR1/2 axis and immune checkpoint inhibitors has demonstrated additional anti-tumor benefits. CXCL8 recruits immunosuppressive cells to the tumor and promotes angiogenesis within the TME [10]. Beyond remodeling the TME, studies have shown that CXCL8 could serve as a prognostic molecular marker in patients undergoing immune checkpoint therapy, and that blocking CXCL8 could improve antitumor efficacy [11].
Currently, CXCL8 expression levels can only be detected in the following ways: peripheral blood cytokine analysis, tissue mRNA quantification, and tissue protein assays (e.g., Western blotting, immunohistochemistry, flow cytometry). These methods are subject to limitations related to the variability of operators and the specificity of antibodies. In contrast, hematoxylin and eosin (H&E) staining is essential for clinical diagnosis and provides the most readily accessible image data. Leveraging artificial intelligence, pathomics converts pathological images into high-fidelity, high-throughput data amenable to extensive analysis. This approach allows the quantification of pathological diagnoses, molecular expression, and disease prognosis using texture, morphological, and biological characteristics [12,13].
This study proposes using pathomics technology to predict CXCL8 expression and prognosis based on HNSCC histopathological images. Additionally, transcriptomics data were integrated to explore the cellular mechanisms underpinning these predictions through pathomics.
MATERIALS AND METHODS
Collection of data cohorts and prognostic analysis of CXCL8
Transcripts per million formatted mRNA-seq data and medical records, including follow-up data, for 528 patients with HNSCC were obtained from the The Cancer Genome Atlas (TCGA)-HNSCC dataset. Using the R package “survminer,” the cutoff value for CXCL8 gene expression for predicting OS was established at 5.372. Clinical information can be found in Table 1. The difference in survival rates between groups with varying levels of CXCL8 expression was analyzed using Kaplan-Meier analysis.
Univariate Cox regression was used to investigate OS influencing factors for correlation analysis and comparison. Multivariate Cox regression was used to identify the independent factor affecting OS, and we also investigated the role of multiple influencing factors.
Histopathological image acquisition, segmentation, and feature extraction
With a maximum magnification of 20× or 40×, H&E‐stained pathology slides were retrieved from the TCGA database [14,15]. The OTSU algorithm (https://opencv.org/) was used to obtain the tissue regions of pathological sections [16]. The 40× images were segmented into multiple subimages of 1,024×1,024 pixels. The 20× image was segmented into multiple subimages of 512×512 pixels, then up-sampled to 1,024×1,024 pixels [15]. The subimages with poor image quality (contamination, image blur, and more than 50% blank area) were reviewed by pathologists and excluded. Twenty subimages were randomly selected from each pathological image for subsequent analysis [14,15]. We used the PyRadiomics open-source package and extracted 93 original characteristics from each sub-image (including the first-order and second-order characteristics). High-order features (Wavelet, LoG, Square, Square Root, Exponential, Gradient, LBP2D) were also extracted. Furthermore, a total of 1,488 features were obtained. After extracting features from 20 subimages of pathological images of each patient, the average value was taken as the pathomic features of each sample for subsequent data analysis [17-19].
There were 313 cases with complete H&E images, gene matrix, and medical records in the TCGA-HNSCC data set. We randomly divided the 313 cases into the sets for training and validation in a ratio of 6:4. Using the z-score, the pathomic feature values of the training set were normalized. The standardization of the validation set depended on the training group’s mean and standard deviation. And the differences in clinical variables among the data sets were analyzed. The workflow of histopathology image processing and analysis pipeline was shown in Supplementary Fig. 1.
Establishment and evaluation of the support vector machines model
At first, the pathomic features were screened by the support vector machines (SVM) model to predict the CXCL8 mRNA level. The efficiency of the SVM model was evaluated in both groups. The evaluation indexes included accuracy (ACC), specificity (SPE), sensitivity (SEN), positive predictive value (PPV), and negative predictive value (NPV). The Brier score was then used to quantify the overall performance of the pathological prediction model. Decision curve analysis (DCA) was used to illustrate the clinical practicability of the pathomics model.
Establishment and evaluation of the logistic regression model
The logistic regression (LR) algorithm was also used to model the features screened by mRMR-RFE. The assessment parameters included ACC, SPE, SEN, PPV, and NPV. The Brier score was used to quantify the overall performance of the pathomics model, and DCA was used to show the clinical practicability of the pathomics prediction model. Furthermore, the fitting status was determined by comparing the area under the curve (AUC) of the training and validation set utilizing the Delong test. The AUC values of the SVM and LR models were also compared.
Correlation analysis of immune genes and immune cell abundance
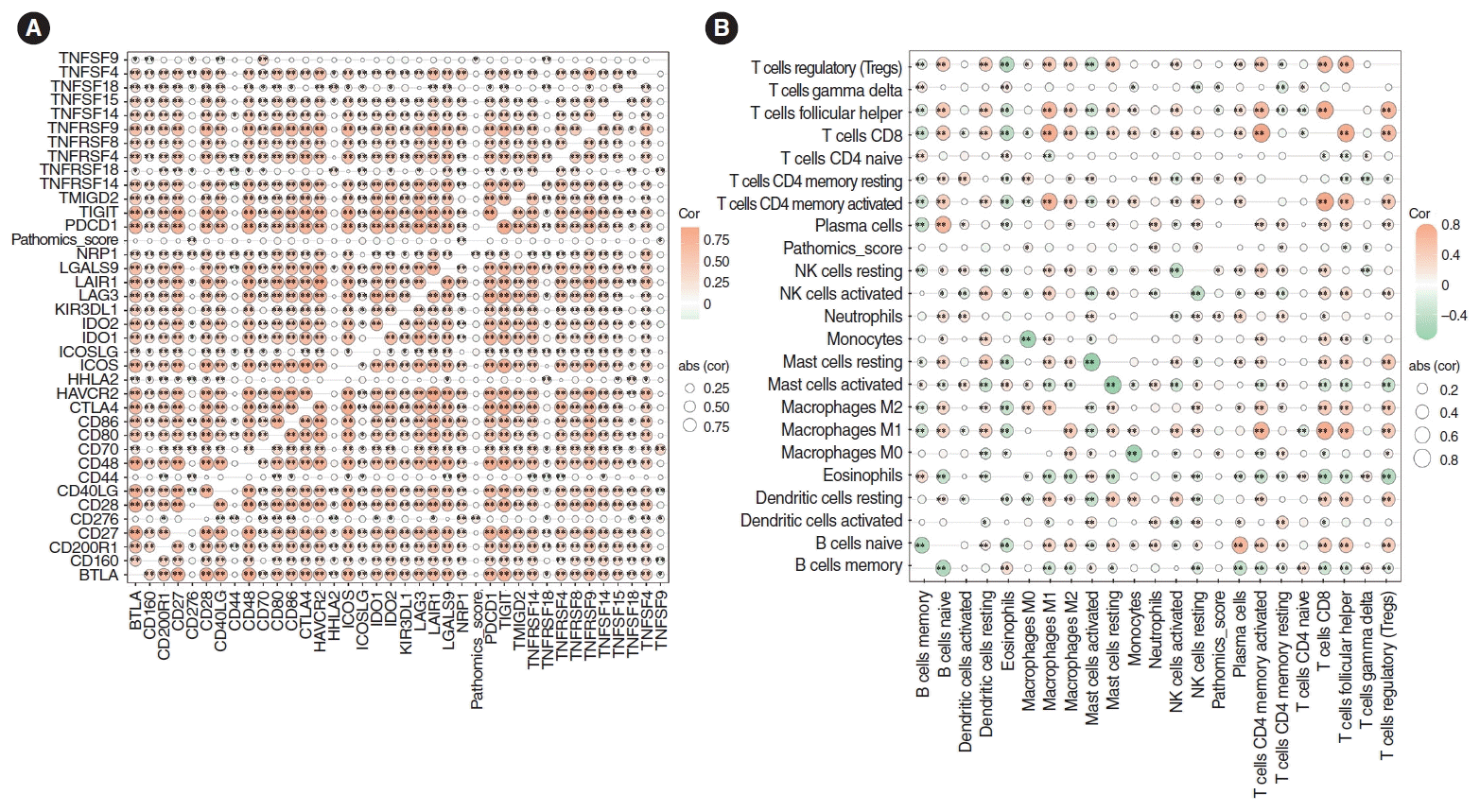
The cut-off value of the pathomics score (PS) of the LR model was set to 0.374, and the cases were then separated into high-PS group (219) and the low-PS group (94). A Spearman correlation analysis was performed between PS and immune-related genes [21]. The gene expression array of the data set was submitted to the CIBERSORTx database, and the association between PS and immune cell infiltration is calculated by the R package “corrplot.”
Model prediction of chemokine differences
The expression differences of chemokine and chemokine receptor-related genes between PS groups were analyzed. R language packages “limma” and “pheatmap” were performed to draw a heatmap. The Wilcoxon rank sum test was then performed to investigate the chemokine and chemokine receptor-related genes between high and low PS sets (gene source; https://www.immport.org/shared/genelists). Moreover, genes with a P <0.05 were visualized through the heatmap display.
Prediction of Gene Set Enrichment Analysis enrichment based on PS
Gene Set Enrichment Analysis (GSEA) enrichment analysis was performed on the differentially expressed genes. The first 20 pathways were visualized by GSEA enrichment analysis in the Kyoto encyclopedia of genes and genomes (KEGG) and Hallmark genes. To explore the molecular mechanisms of differential expression genes between PS groups, R package “clusterProfiler” was performed on KEGG (c2. Cp. KEGG. 7.5.1. symbols. gmt) gene sets and Hallmark (h.all. 7.5.1. Symbols. gmt) gene sets to analyze GSEA enrichment (P <0.05).
Correlation analysis of chemokines and chemokine receptors
The R package “WGCNA” was used to search for chemokine and chemokine receptor gene sets related to prognosis, and the expression similarity matrix was constructed according to the expression data of chemokines and chemokine receptors. The power value of scale independence and average connectivity of networks was converted into a topological matrix, and we utilized the topological overlap metric (TOM) to describe the correlation degree among genes. The genes were clustered with 1-TOM as the distance, and the clipping height was set at 0.4 for dynamic clipping module identification. At last, modules with the highest correlation with clinical features were recognized as the key module, and then the relevant hub genes were identified.
Gene ontology and KEGG enrichment analysis
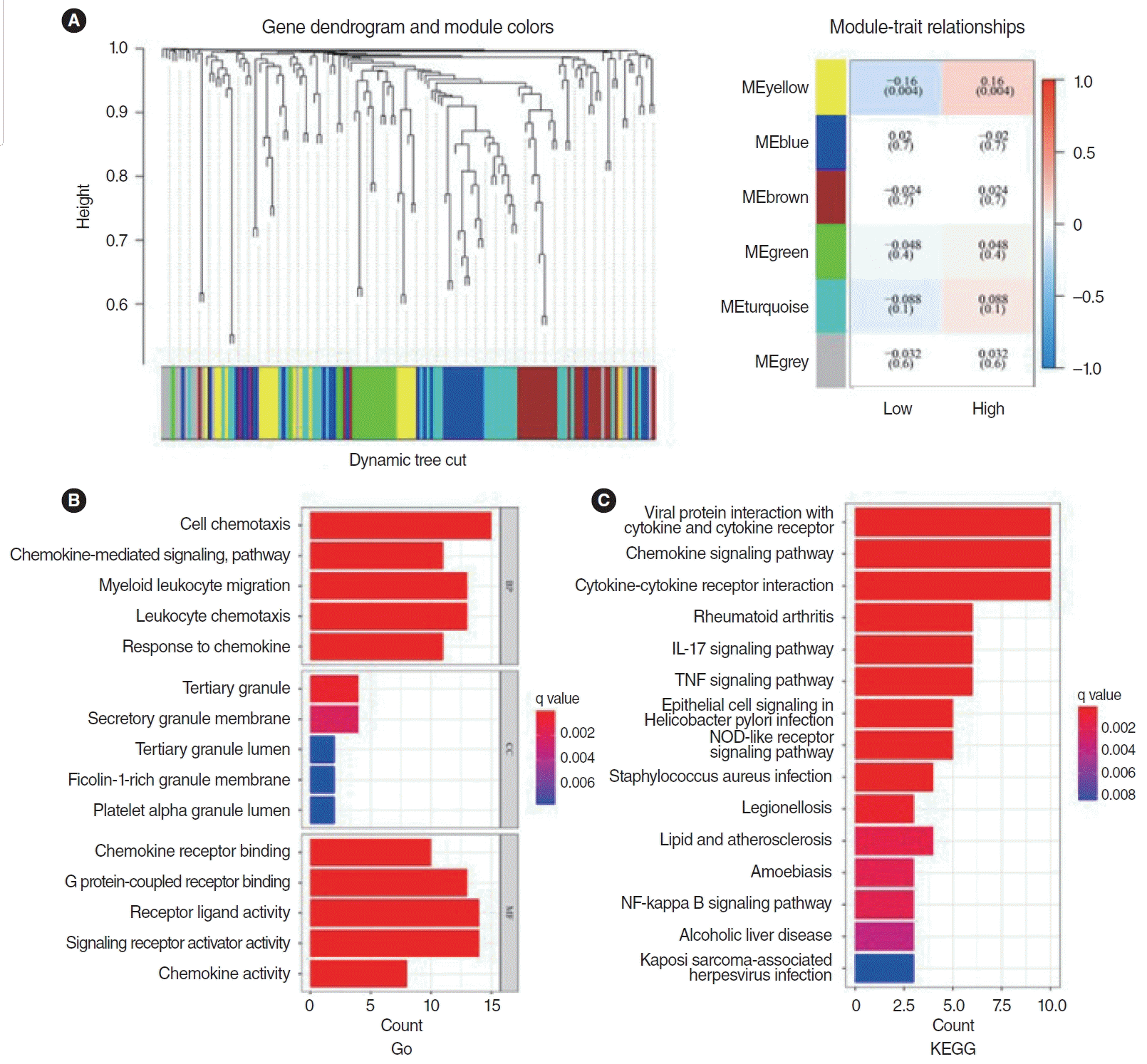
To further confirm the potential functions of the 20 chemokines and chemokine receptor-related hub genes in the yellow module, we analyzed the data using functional enrichment. KEGG enrichment analysis was employed to visualize the molecular functions (MF), biological processes (BP), and cellular components, highlighting the top 15 significantly enriched pathways. The top five significantly enriched pathways were visualized for MF. The R package “clusterProfiler” was used for enrichment analysis, considering a statistical significance threshold of P <0.05.
Statistical analysis
Categorical variables were presented as frequencies and percentages (%), with clinical features assessed using either Fisher’s exact test or the chi-squared test. For non-normally distributed continuous variables, group comparisons were performed using the Wilcoxon test. The Log-rank test was employed to evaluate the significance of survival rates across groups, defining median survival time as the duration corresponding to a 50% survival rate. Baseline data comparison utilized the R package “CBCgrps,” while survival analysis and visualization were executed using the “survival,” “cmprsk,” and “forestplot” packages. Furthermore, the “clusterProfiler” and “stats” packages facilitated the investigation into the biological mechanisms underlying pathological features and diseases. All statistical analyses were carried out using R software version 4.1.0, adopting a two-sided hypothesis testing approach, with P<0.05 considered statistically significant.
RESULTS
Prognostic analysis of CXCL8
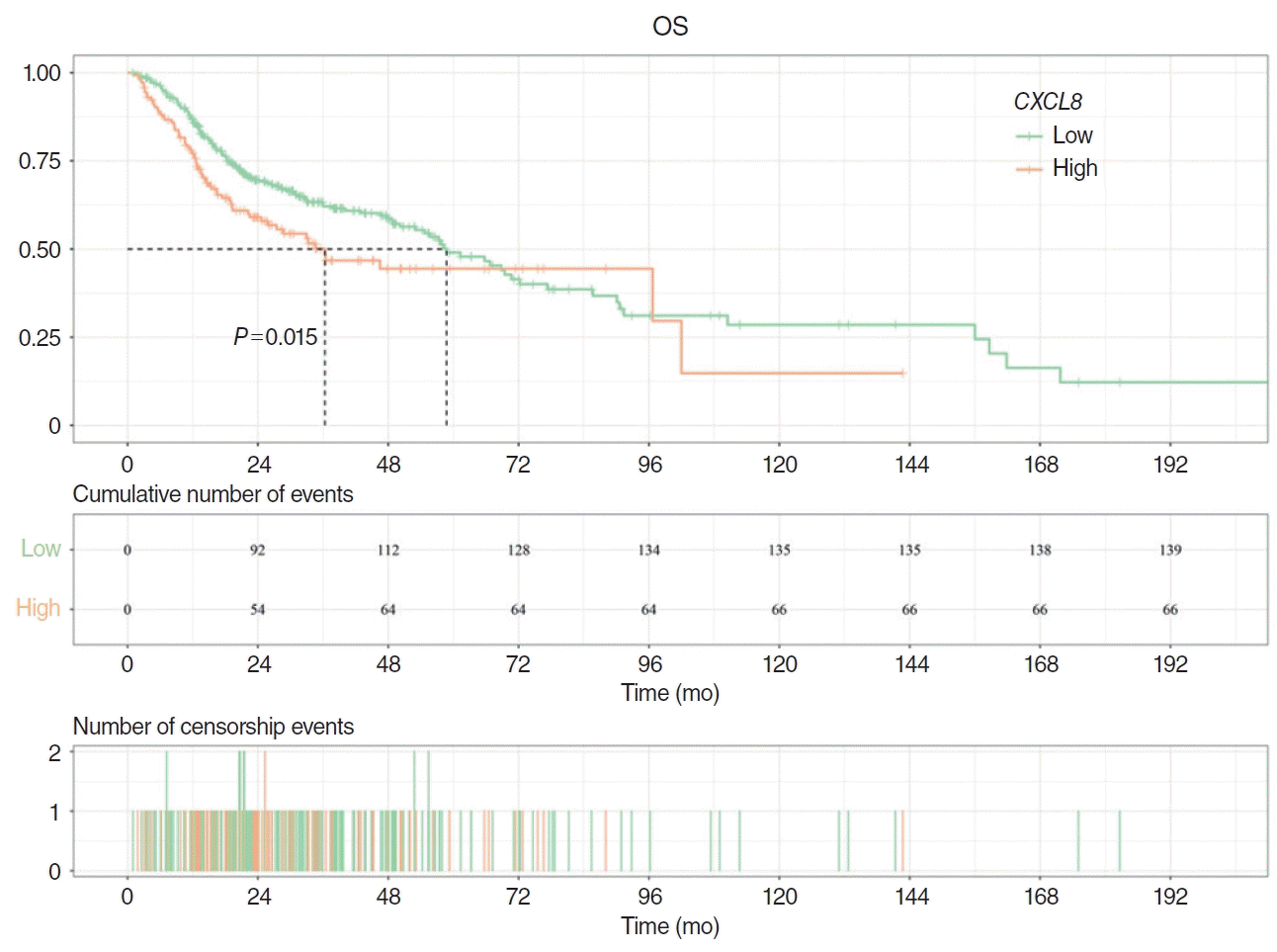
The correlation between CXCL8 mRNA levels and clinical characteristics in the HNSCC dataset is presented in Table 1. CXCL8 expression was significantly associated with HPV status (P<0.05). Patients in the low CXCL8 expression group exhibited a substantially longer median survival time compared to those in the high expression group (58.73 months vs. 36.33 months). Additionally, CXCL8 expression in tumor tissue was higher than in normal tissue (P <0.05). Kaplan-Meier curves also demonstrated a significant association between high CXCL8 expression and shortened OS (P =0.015) (Fig. 1).
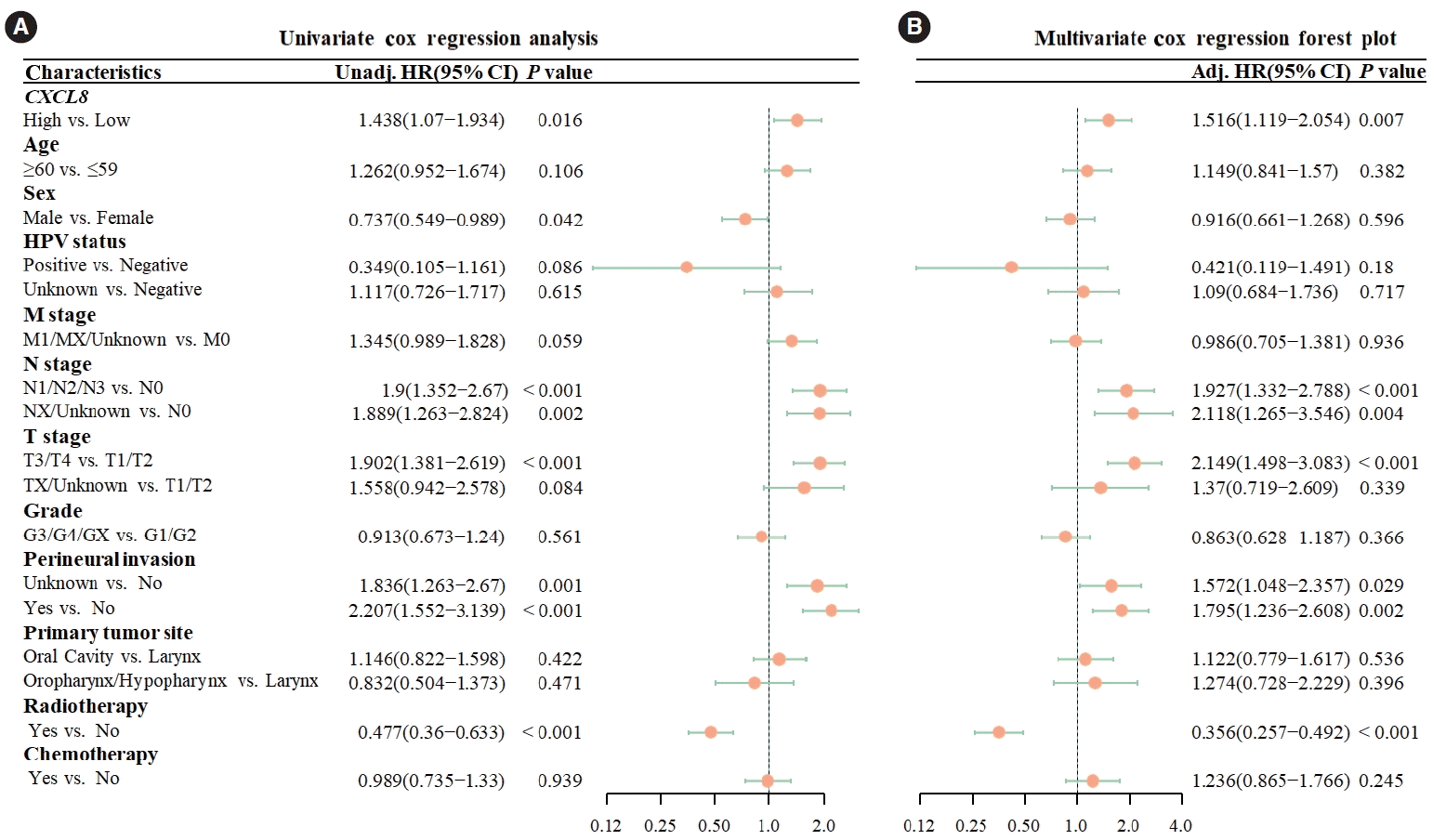
Univariate Cox analysis revealed that increased CXCL8 expression was a risk factor for OS (hazard ratio [HR], 1.438; 95% confidence interval [CI], 1.07–1.934; P =0.016). After multivariate adjustment, high CXCL8 expression continued to be a significant risk factor for OS in the multivariate Cox analysis (HR, 1.492; 95% CI, 1.103–2.019; P =0.009) (Fig. 2).
Histopathological image acquisition, segmentation, and feature extraction
This study rigorously adhered to the inclusion and exclusion criteria for patient selection, segmenting the data into a training set (n=189) and a validation set (n=124) at a 6:4 ratio. Utilizing the training set, we developed a pathomics model, which was subsequently validated with the validation set. Patients in both the training and validation sets had similar baseline conditions (P >0.05) (Table 2).
mRMR-RFE feature screening
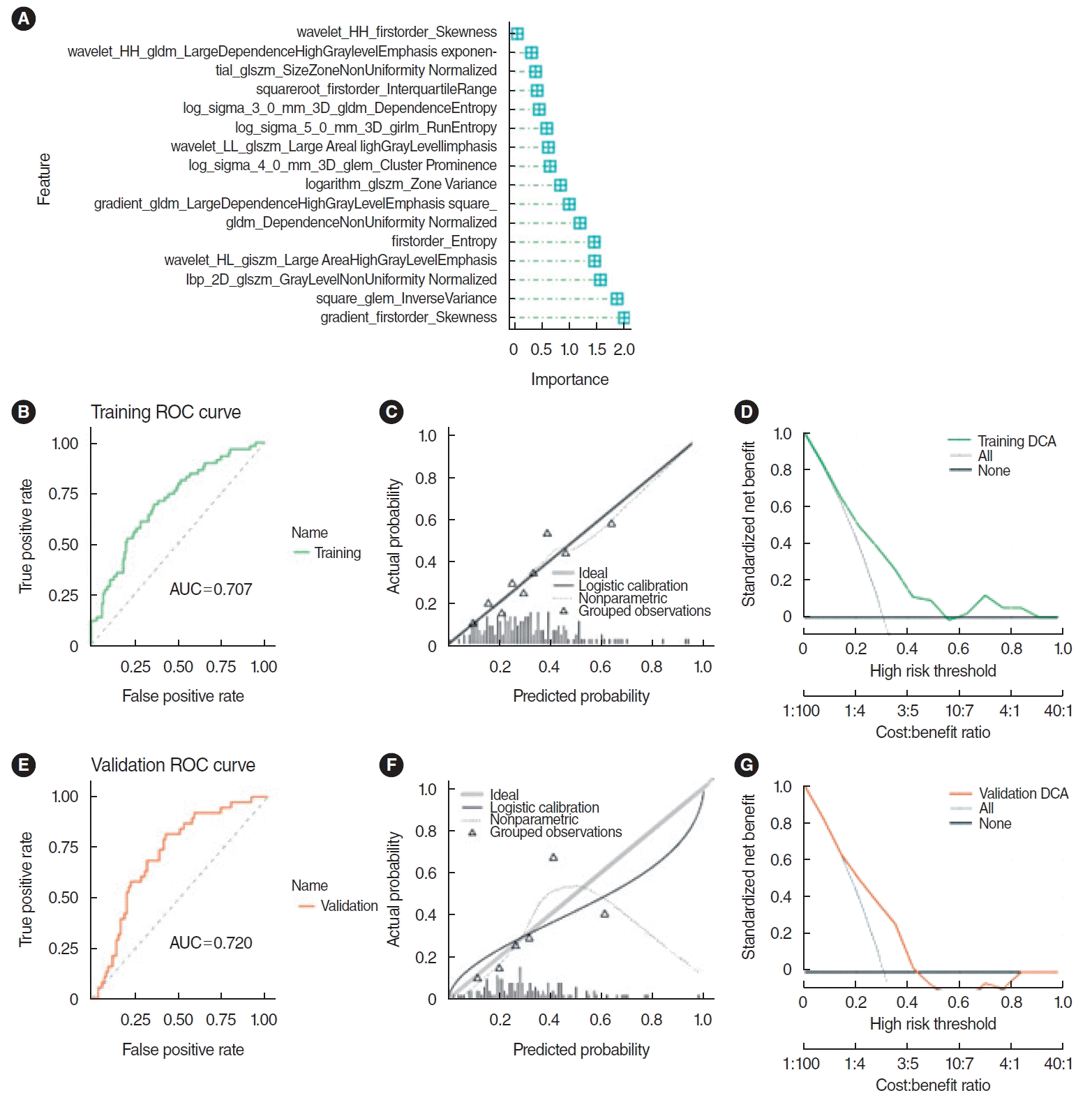
The top 30 features were screened by the mRMR method and then selected by RFE. Sixteen features were obtained through mRMR-RFE screening (Fig. 3).
Establishment and evaluation of the SVM model
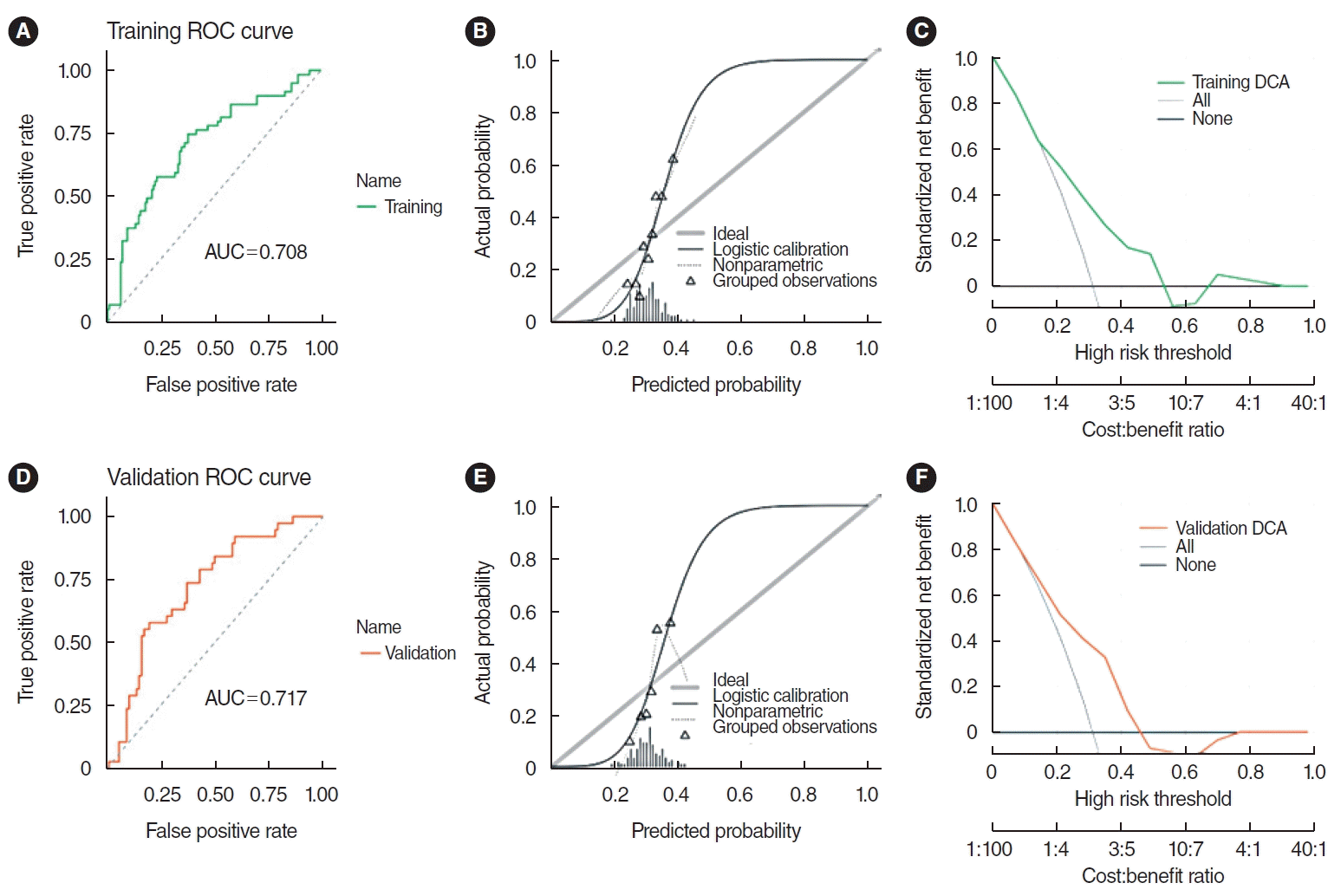
The SVM model demonstrated robust predictive ability. The AUC for the training set was 0.708, while the validation set achieved an AUC of 0.717, as illustrated by the receiver operating characteristic (ROC) curve. Furthermore, the AUC values for both sets did not differ significantly (P=0.881), indicating a satisfactory model fit. However, the calibration curve showed a poor correlation between the predicted probability of high gene expression levels in the pathomics model and the actual observed values (P <0.05 for the Hosmer-Lemeshow test). DCA indicated that the model has good clinical applicability (Fig. 4).
Establishment and evaluation of the LR model
The LR model demonstrated excellent predictive power. The AUC value for the training set was 0.707, and that for the validation set was 0.72, as indicated by the ROC curve. There was no significant difference between the training and validation set AUC values of the LR model (P =0.912), indicating a good model fit. The calibration curve showed that the predicted probability of high gene expression by the pathomics prediction model closely matched the actual observed values (P >0.05). DCA indicated that the model was highly suitable for clinical use (Fig. 5). The PS of the training set revealed differences between the two gene expression groups (P <0.001), with the group exhibiting higher CXCL8 expression having an elevated PS in both the training and validation sets. The AUC values of the LR model were comparable to those of the SVM model in both sets (P =0.944 and P =0.849, respectively), and both models showed good predictive performance. However, unlike the SVM model, the HL test P-value for the LR model was >0.05, indicating a good fit with the actual observed values. Consequently, the predictive results of the LR model were selected for further analysis.
Correlation analysis of immune genes and immune cell abundance
Spearman correlation analysis was conducted, revealing that the PS was positively correlated with immune-related genes TNFSF9 (P <0.05), CD276, and NRP1 (P <0.01). Additionally, the infiltration of immune cells in the HNSCC dataset was analyzed, demonstrating a positive correlation between PS and the degree of neutrophil cell infiltration (P <0.05) (Fig. 6).
Correlation analysis of chemokines and chemokine receptors
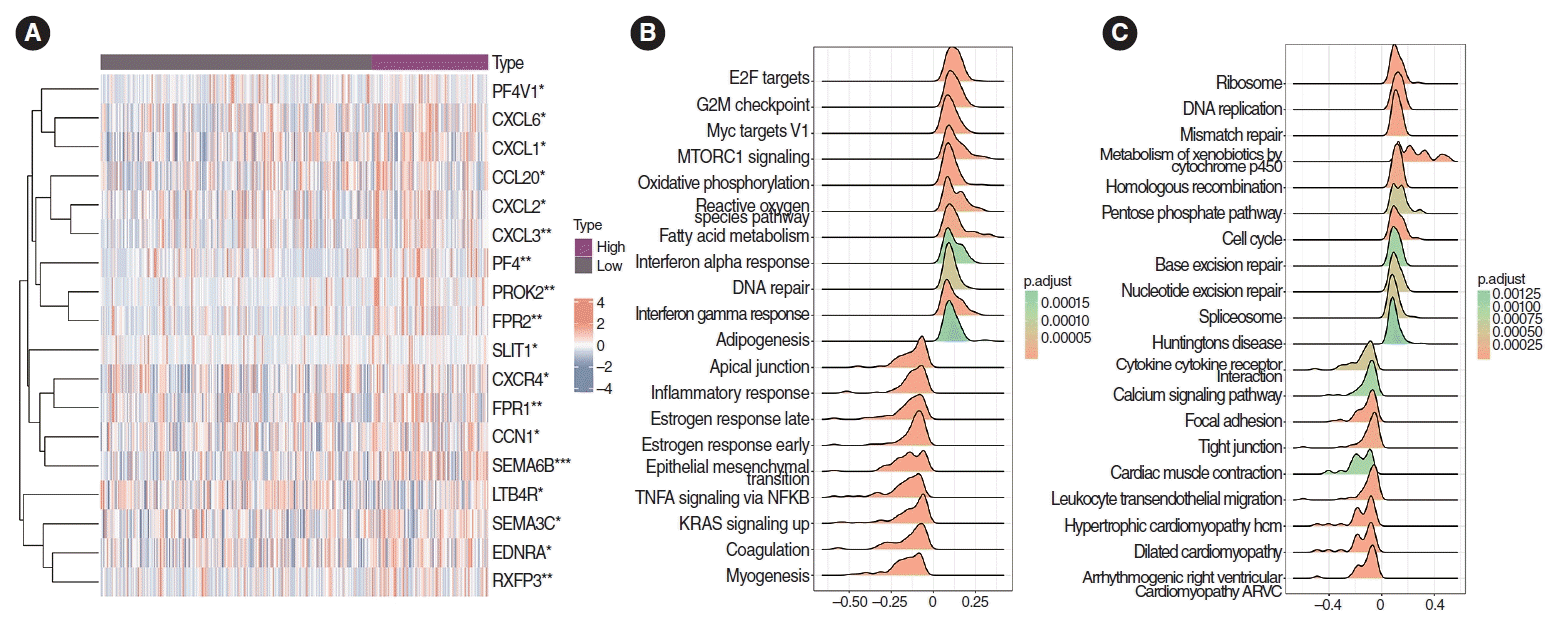
SEMA6B (P<0.001), CXCL3, PF4, PROK2, and FPR1 (P<0.01) expression levels were higher in the high-PS set than in the low-PS set (significance mark: NS, P≥0.05, *P<0.05, **P<0.01, and ***P <0.001) (Fig. 7A).
Prediction of GSEA enrichment based on PS
In the Hallmark gene set, GSEA enrichment analysis visualized the top 20 pathways, and the differentially expressed genes in the high-PS group were significantly enriched in G2M checkpoint and DNA repair pathways (Fig. 7B). The low-PS group showed significant enrichment of Kirsten Rat Sarcoma Viral Oncogene Homolog and inflammatory response pathways.
In the KEGG gene set, GSEA enrichment analysis visualized the top 20 pathways, and the cell cycle pathway was significantly enriched in the high-PS group. Meanwhile, in the low-PS group, cytokine-cytokine receptor interaction and leukocyte transendothelial migration pathways were significantly enriched (Fig. 7C).
Correlation analysis of chemokines and chemokine receptors
The optimal soft threshold power for the scale-free network was determined to be 3. Subsequently, six modules were selected by clustering the average connectivity hierarchy with this optimal soft threshold power. Each module was assigned 154 genes. Among these, the yellow module exhibited a strong association with the prognosis of HNSCC, as indicated by the Pearson correlation coefficient between the modules and the sample characteristics of each module. Consequently, the yellow module was identified as the key module (Fig. 8A).
Gene ontology and KEGG enrichment analysis
The gene ontology (GO) enrichment analysis suggested that the 20 hub genes in the yellow module are enriched in pathways related to cell chemotaxis, chemokine-mediated signaling, myeloid leukocyte migration, leukocyte chemotaxis, and response to chemokines (Fig. 8B). Similarly, KEGG enrichment analysis indicates that these hubs are significantly enriched in pathways involving cytokines and their receptors, chemokine signaling, cytokine-cytokine receptor interaction, and tumor necrosis factor (TNF) and interleukin (IL)-17 pathways (Fig. 8C).
DISCUSSION
Pathomics enables the quantitative analysis of neoplasms by examining numerous pathological characteristics, thereby quantitatively assessing tumor heterogeneity. As such, pathomics holds potential clinical value in predicting cancer prognosis and enhancing therapeutic strategies [17,22-24]. The field of pathomics offers broad application prospects and can facilitate a deeper understanding of the TME, paving the way for new directions in basic research. Recent studies indicate that tumor-derived CXCL8 signaling can promote an immunosuppressive TME [25,26]. Randomized studies have demonstrated that increased levels of CXCL8 are associated with a reduced benefit from immune checkpoint inhibitors [27,28]. In colorectal cancer, CXCL8 is closely associated with the TME and the response to immunotherapy, affecting immune evasion and potentially improving the prognosis of malignant glioma, as well as the efficacy of chemotherapy [5]. For breast cancer patients, higher levels of CXCL8, along with CXCL10 and CXCL11, have been linked to decreased OS, emphasizing its potential as both a therapeutic target and a prognostic marker in breast tumors [6]. The role of CXCL8 as a potential biomarker for gastric cancer is noteworthy; its upregulation in gastric cancer tissues correlates with shorter relapse-free survival, underscoring its significant role in disease progression and prognosis [7]. In recurrent ovarian cancer, CXCL8 shows increased expression, while its expression diminishes in late-stage tumors, suggesting its utility as a diagnostic biomarker [8]. Additionally, in esophageal cancer patients, elevated CXCL-8 levels correlate positively with the depth of tumor invasion and CRP concentrations, further supporting its potential as a tumor marker [9]. Overall, the significance of CXCL8 in the TCGA data is confirmed by its substantial impact on various cancer types, reinforcing its importance as a biomarker for disease progression, diagnosis, and prognosis.
This study integrated multiple data perspectives, including clinical features, transcriptomics, and pathomics, to analyze HNSCC. Initially, we identified CXCL8 as a prognostically significant hub gene in HNSCC patients. In line with prior studies, we found that patients with higher levels of CXCL8 expression in tumor tissues had a worse prognosis, as indicated by data from the TCGA HNSCC dataset [29-31]. Recognizing the meaningful link between CXCL8 expression and OS, we then employed pathomics features to assess CXCL8 expression levels. For this assessment, 16 pathomics features were selected using the SVM method, and the resulting model demonstrated robust performance in both training and testing cohorts. Histopathological image features proved capable of evaluating CXCL8 expression levels. In related work, Wang et al. [32] developed an SVM model with RFE to predict CD27 expression. Their model’s predictive probability was positively correlated with gene expression, and DCA confirmed its high clinical utility. No significant differences were found between the AUC values of the training and validation sets. In our study, however, the calibration curve of the SVM model indicated a poor match between the model’s predicted probabilities and the actual values. To address this, we developed an LR model that showed better consistency than the SVM model. We subsequently used the LR model for further analysis.
Previous studies have demonstrated that CXCL8 signaling can facilitate the recruitment of neutrophils to the TME, which may promote tumor growth [33] and immune resistance [34]. The correlation analysis of immune cell abundance in the current study showed that PS expression was positively correlated with neutrophil infiltration, aligning with prior findings.
KEGG and GO enrichment analyses were conducted to investigate the underlying biological processes. The Hallmark gene set GSEA enrichment analysis highlighted the top 20 pathways, revealing that the differential genes in the high-PS group were predominantly enriched in DNA repair and G2M checkpoint pathways, suggesting reduced DNA damage. GO enrichment analysis indicated that the high PS expression group showed significant enrichment in processes such as cell chemotaxis, myeloid leukocyte migration, leukocyte chemotaxis, and response to chemokines. KEGG enrichment analysis revealed an upregulation in cytokine-cytokine receptor interaction, chemokine signaling pathways, and TNF and IL-17 pathways. Previous research has also established a link between immune-related pathways and the morphological features of lymphocytes and cancer cells [14]. These insights contribute to our understanding of the molecular mechanisms related to the morphological characteristics of tumor cells.
The CXCL8-derived pathomics model demonstrated effectiveness in predicting the prognosis of HNSCC in our study; however, it is important to recognize its limitations. First, while the model was constructed and validated using a large cohort from the TCGA databases, the study is inherently retrospective. To confirm the model’s validity, a prospective study involving a multi-center patient cohort is essential. Second, although CXCL8 dysfunction has been linked to immune checkpoints in several studies [35], further in vivo and in vitro research is necessary to elucidate the mechanisms by which CXCL8 influences immune checkpoints. Third, in our study, experienced pathologists manually delineated the regions of interest, a process that is not only labor-intensive but also prone to interobserver variability. Future studies should aim to validate our findings using multicentric prospective research and incorporate more advanced and efficient tumor segmentation algorithms.
In this study, we developed and validated a pathomics signature using machine learning methods. CXCL8 may serve as a predictor of survival for patients with HNSCC, thus assisting in clinical decision-making. The pathomics features of CXCL8 could be instrumental in exploring the mechanisms through which CXCL8 impacts HNSCC patients. Analyzing features from histopathological images may offer a feasible and economical method for predicting molecular expression levels in patients with HNSCC.
HIGHLIGHTS
▪ The study introduces an innovative pathomics model that utilizes machine learning techniques on hematoxylin and eosin stained images to precisely predict CXCL8 expression in head and neck squamous cell carcinoma (HNSCC), demonstrating its potential to enhance patient survival rates via personalized therapy approaches.
▪ The research illustrates the model’s effectiveness in stratifying HNSCC patients according to survival outcomes, recognizing high CXCL8 expression as a crucial prognostic indicator for poor prognosis, thereby enabling more precise therapeutic interventions.
▪ The study integrates clinical, transcriptomic, and pathomics data into a multidimensional model, validated against The Cancer Genome Atlas datasets, to improve prognostic accuracy and advocate for the integration of quantitative image analysis in personalized treatment strategies for HNSCC patients.

 XML Download
XML Download