

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Prolonged jaundice lasting longer than 21 days of life is not an uncommon phenomenon in infants in the aspect of neonatologists. It occurs in approximately 15% of all newborn infants [1,2]. Although some patients may have hypothyroidism or liver dysfunction as an underlying cause requiring medical or surgical intervention, most other infants have benign unconjugated hyperbilirubinemia, and no specific results have been obtained after routine investigation for prolonged jaundice. The prognosis of unconjugated hyperbilirubinemia is usually good, and bilirubin levels improve within a few weeks if the patient receives the recommended standard of care [3]. It is necessary for physicians to pay medical attention to prevent prolonged hyperbilirubinemia and resulting bilirubin-induced neurological damage. Primary care providers should monitor the cause of these symptoms if jaundice persists beyond a typical time frame.
Uridine diphosphate glucuronosyltransferase 1A isoform 1 (UGT1A1) is a key enzyme that helps eliminate bilirubin from the body by adding a glucuronide moiety to substrates [4,5]. Mutations in UGT1A1, which is located on chromosome 2q37, can result in hereditary unconjugated hyperbilirubinemia, specifically Crigler-Najjar syndrome (CN) and Gilbert’s syndrome (GS), owing to varying degrees of enzymatic activity reduction [6-11]. CN is a rare genetic disorder characterized by a complete (or nearly complete) absence of the ability to eliminate bilirubin from the body by glucuronidation. This disorder has two forms, depending on the degree of elevated bilirubin concentration and reduced UGT1A1 enzyme activity: CN type 1 and type 2. Mutations in CN type 1 cause severe, life-threatening hyperbilirubinemia with an almost complete absence of UGT1A1 enzymatic activity, whereas mutations in CN type 2 cause a moderate, less severe form of unconjugated hyperbilirubinemia with a reduced level of enzymatic activity [12-14]. In comparison, GS is a little more common inherited disorder presenting as a mild form of unconjugated hyperbilirubinemia with approximately 30% reduced UGT1A1 enzymatic activity [15,16]. The prevalence of GS is approximately 4% to 14% of the global population, with one in three affected patients unaware that they have it [17-19].
To date, more than 100 genotypes of polymorphism of UGT1A1 have been identified from different countries around the world. They are usually found in the promoter or coding exon regions [11,15,20,21]. These mutations have been variably associated with GS and CN and appear to differ considerably among ethnic groups [22,23]. For example, the insertion of an extra TA into the normal sequence of the promoter TATA box, (TA)7 TAA (c.-40_-39dupTA or c.-40_-39 insTA), is a common genotype related to GS in Caucasians [8,22,24], whereas p.Gly71Arg (c.211G>A), a missense mutation in the coding exon 1 region, is more prevalent in Asian populations [10,15,25,26].
Other populations have been extensively studied; however, research on UGT1A1 mutations in Koreans remains limited and requires further investigation. Previous studies in Korea have focused mainly on adult patients or infants with small sample sizes and examined a few specific single-nucleotide variants [27-31]. To the best of our knowledge, there are no national statistics on the prevalence of UGT1A1 mutations in Korean infants with prolonged jaundice.
This study aimed to identify the types and prevalence of UGT1A1 mutations in newborn infants with unexplained prolonged unconjugated hyperbilirubinemia and determine the clinical relationship among infants with different mutational variations of UGT1A1.
MATERIALS AND METHODS
1. Study participants
The study included 74 infants who visited the neonatal jaundice follow-up clinic of Samsung Changwon Hospital from July 2019 to May 2023. All enrolled infants underwent a thorough examination to identify the underlying cause of prolonged jaundice, including measurement of the peak levels of total and direct serum bilirubin, complete blood count, peripheral blood smear, reticulocyte count, Coombs test, liver function test, thyroid function test, and liver ultrasonography. Mutation analysis of UGT1A1 was conducted in patients whose parents consented. In this study, unexplained prolonged jaundice was defined as jaundice that persisted beyond 21 days of life in infants who demonstrated no evidence of hemolytic anemia, liver dysfunction, hypothyroidism, or inborn errors of metabolism [32]. Peak levels of total and direct bilirubin were defined as the highest levels of total and direct bilirubin measured at 21 days of life or later. Patients with gestational age <35 weeks, birth asphyxia, dehydration, cephalohematoma, intracranial hemorrhage, sepsis, immune hemolytic anemia, glucose-6-phosphate dehydrogenase (G6PD) deficiency, medications associated with jaundice, hypothyroidism, hepatobiliary disease, and inborn errors of metabolism were excluded from the study.
2. Clinical and laboratory data collection
Clinical and laboratory information of the participants was retrospectively reviewed from hospital medical records. Recorded data included demographics (age, sex, date of birth, gestational age, and birth weight), delivery method, breast milk/formula, stool pattern/color, blood type, maternal/family history of jaundice, serum levels of total and direct bilirubin, peak levels of bilirubin, and the presence or absence of liver dysfunction and hypothyroidism. The findings of the mutation analysis of UGT1A1 were also retrieved.
3. Genetic studies
For sequence analysis of UGT1A1 gene mutation, the blood samples from the participants were withdrawn and requested from the Department of Laboratory Medicine and Genetics of Samsung Medical Center, Seoul, Korea. Written informed consent for mutation analysis was obtained from the patient’s parents before collecting the specimens.
Genomic DNA was extracted from peripheral blood leukocytes using a Wizard Genomic DNA Purification Kit (Promega) according to the manufacturer’s instructions. The promoter region and five exons of UGT1A1 were amplified by polymerase chain reaction (PCR). PCR was performed using a thermal cycler (Applied Biosystems). The PCR products were purified, and direct sequencing was performed using an ABI3730xl Genetic Analyzer (Applied Biosystems). The obtained sequences were compared to the reference sequences NG 002501.2 and NM_000463.2, registered in the National Center for Biotechnology Information database (http://www.ncbi.nlm.nih.gov).
4. Data analysis
Statistical analyses were performed using SPSS Statistics for Windows version 23.0 (IBM Corp.). Numerical data were expressed as mean±standard deviation and analyzed using Student’s t-test and Mann-Whitney test. Chi-square, Fisher’s exact, and likelihood ratio tests were used to compare the categorical data, such as sex, delivery method, and breastfeeding. Differences were considered statistically significant at P<0.05.
RESULTS
1. Demographics
A total of 65 infants with prolonged unconjugated hyperbiliruPercent binemia were enrolled in this study after excluding nine infants for the following reasons: two infants had biliary atresia, one had hypothyroidism, one had cytomegalovirus hepatitis, and five lacked clinical information. None of the enrolled infants showed evidence of hemolytic anemia, liver dysfunction, hypothyroidism, or inborn metabolic errors. No abnormalities, including hemangioma, biliary atresia, or hepatic parenchymal disease, were found on liver ultrasonography. Mutation analysis of UGT1A1 was performed in 33 (50.8%) patients whose parents consented to participate.
2. UGT1A1 genotypes
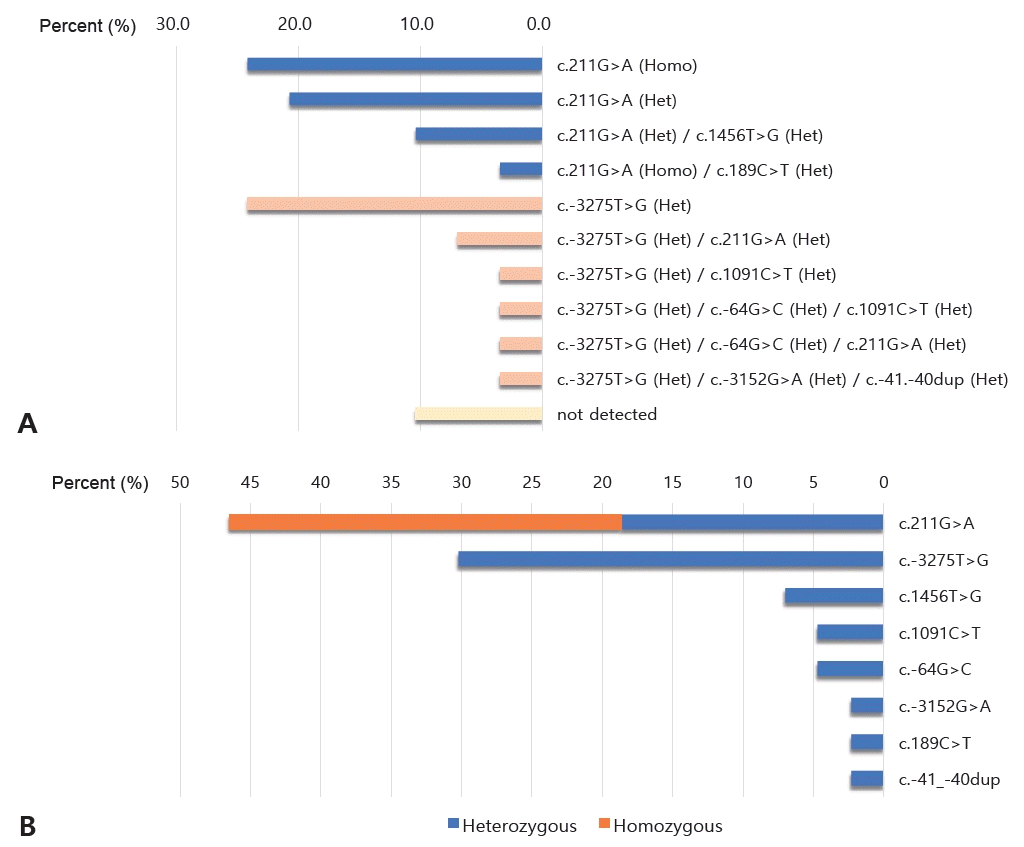
Among the participants who underwent mutation analysis, 30 (90.9%) harbored mutations in UGT1A1. Single-nucleotide variants were found in 20 (66.7%) infants, and the remaining 10 (33.3%) had multiple variants of UGT1A1. Eight different genetic variants were detected by the mutation analysis of UGT1A1 (Figure 1A). The two most common variants were c. 211G>A (46.5 %) and c.-3275T>G (30.2%) (Figure 1B). The other variants had the following percentages: c.1456T>G (7.0%); c.-64G>C and c.1091C >T (4.7% each); and c.-3152G>A, c.189C>T, and c.-41_-40 dup (2.3% each). Among the 20 infants with the c.211G>A variant, eight (40.0%) had a homozygous genotype and 12 (60.0%) had a heterozygous genotype. Infants harboring other variants exhibited heterozygous genotypes. When comparing the group that underwent mutation analysis with the group that did not, there were no significant differences in demographics.
3. Effects of UGT1A1 genotypes on clinical and laboratory parameters of patients
Clinical and laboratory data were assessed to identify differences among groups with varying UGT1A1 mutations. When analyzing the patient data between the groups in which UGT1A1 mutation was detected (n=30) and not detected (n=3), it was found that breastfeeding was the only significant factor (P=0.027). All infants with prolonged unconjugated hyperbilirubinemia without UGT1A1 mutations were exclusively breastfed, whereas less than one-third of the infants with confirmed mutations were breastfed (Table 1). There were no significant differences in the total and peak bilirubin levels between the two groups.
No significant differences were observed between the group with a single-nucleotide variant and the group with multiple genetic variants, or between the homozygous genotype group with c.211G>A and the heterozygous genotype group. No significant differences were observed between the groups of infants with the two most common variants, c.-3275T>G and c.211G>A (Table 2).
DISCUSSION
This study demonstrates that infants with prolonged unconjugated hyperbilirubinemia are highly likely to have a UGT1A1 mutation. This unexpected result deviates from our initial expectations. As we were unsure, at the beginning of this study, how many participants would be experiencing prolonged jaundice with genetic backgrounds such as GS or CN, the mutation analysis of UGT1A1 gene was reluctantly recommended and performed only for those who requested it, seeking the possible underlying disease. As a result, just about half of the recruits received the UGT1A1 gene mutation analysis. Although the number of participants who underwent mutation analysis was small and limited, this result suggests that a substantial number of newborns who experience prolonged jaundice lasting longer than 3 weeks of life may suffer from hereditary unconjugated hyperbilirubinemia. Therefore, UGT1A1 mutation analysis may be a reliable option for determining the underlying cause of prolonged jaundice, especially if routine clinical and laboratory investigations, including liver and thyroid function tests, yield no specific results.
This study also demonstrated that c.211G>A in the coding region and c.-3275T>G in the promoter region were the two most common polymorphisms in infants with prolonged unconjugated hyperbilirubinemia. These results are consistent with reports from other Asian populations, including Japanese, Chinese, and Vietnamese [2,32-34]. However, the prevalence of TA duplications in the TATA box promoter was low in the present study. Previous studies have reported that the frequency of polymorphisms in UGT1A1 and their association with neonatal hyperbilirubinemia varies among populations by location, race, and ethnicity [8,25,35,36]. For example, the frequency of the c.211G>A mutation is higher in Asian patients with neonatal hyperbilirubinemia than in European or African individuals. In contrast, mutations in the (TA)n tandem repeat sequence within the TATA box promoter region are more prevalent in Caucasians and Africans than those in East Asians. Kim et al. [31] reported that c.211G>A and c.-41_-40 dup TA were the most frequent variants in 81 Korean patients with unconjugated hyperbilirubinemia. The prevalence of TA duplication was higher than that in data from Japanese and Chinese populations, which was also different from the results of this study. Therefore, more participants are needed to clarify the popularity of polymorphisms of UGT1A1 mutations in the future.
The exact etiology of breast milk jaundice has not yet been determined. Most of the proposed etiologies involve factors present in human breast milk. These factors, including pregnane-3α, 20β-diol, interleukin 1β, β-glucuronidase, epidermal growth factor, and α-fetoprotein, are believed to enhance intestinal resorption of bilirubin and reduce intestinal motility in the neonatal period, leading to increased unconjugated bilirubin levels [37,38]. Other hypotheses suggest the presence of potential genetic mutations in the affected neonates. Studies have reported different results regarding the prevalence of UGT1A1 mutations in infants with prolonged jaundice. Maruo et al. [39] reported that 16 of 17 breastfed Japanese infants with prolonged jaundice had at least one UGT1A1 mutation. Yang et al. [40] also reported that 58.9% of Chinese patients with prolonged jaundice harbored the c.211G>A variant of UGT1A1, which was significantly higher than that in the control group. Alaee et al. [41] reported no significant association between prolonged Iranian neonatal jaundice and UGT1A1 G71R polymorphism. Therefore, more studies with a larger number of participants are needed to reveal the prevalence of UGT1A1 mutations in infants with prolonged jaundice.
In previous studies, the serum bilirubin levels in the homozygous polymorphism subgroup were significantly higher than those in the wild-type and heterozygous subgroups. This suggests that homozygous UGT1A1 mutation carriers may have a remarkable decrease in the enzymatic activity of UGT1A1, resulting in a more elevated serum bilirubin leve [l8,32,42]. Bosma et al. [8] reported that individuals with homozygous (TA)7 TA duplications had higher serum bilirubin levels than those with heterozygous or wild-type alleles. Nguyen et al. [32] also noted that the serum bilirubin level in the homozygous c.211G>A polymorphism subgroup was significantly higher than those in the wild-type and heterozygous c.211G>A polymorphism subgroup. It is likely that the heterozygous polymorphism only partially impaired the enzymatic activity of UGT1A1, and its influence on the conjugation of indirect bilirubin was not noticeable. However, contrary to studies in other countries, we could not find differences in peak total and direct bilirubin levels between the homozygous and heterozygous c.211G>A polymorphism groups. Identical results were obtained when groups with different divisional criteria were compared. This may be partly because our data on peak bilirubin levels were collected from patients beyond 21 days of life and not during their entire lifetime after birth. There might have been a difference if the bilirubin levels were measured consecutively immediately after birth or once after the patient’s symptoms appeared. Again, the number of participants and time to measure the bilirubin levels should be upgraded to interpret their clinical significance correctly.
We did not differentiate between the GS and CN groups in this study. Usually, the distinction between CN type 1 or 2 and GS is based on the severity of serum bilirubin levels, UGT1A1 enzymatic activity, and responsiveness to phenobarbital administration in neonatal jaundice [43]. However, it is challenging to differentiate between them in clinical practice because many patients in this study had a similar range of serum bilirubin levels (intermediate between the two syndromes) measured after 3 weeks of life. All these forms of hereditary unconjugated hyperbilirubinemia are caused by mutations in UGT1A1. They are considered part of the spectrum of a single disorder with varying grades of severity and altered glucuronidation that can extend to fatal CN [44-46]. Nevertheless, all participants in this study were monitored and followed for the persistence of jaundice, and their symptoms gradually disappeared by approximately 8 to 10 weeks of age without any sequelae.
As the prognosis is generally good, GS does not require dietary therapy or specific management. However, mutation analysis of UGT1A1 could not only confirm the diagnosis but also alleviate patients’ parents’ concerns and provide genetic counseling to prevent repeated examinations or hospitalization of patients. Although no specific therapy is required for patients with GS, they should be educated to avoid exacerbating conditions and increasing the risk of toxicity with specific medications. For example, irinotecan, an anticancer drug used to treat colon and small cell lung cancers, can increase the risk of toxicity in patients with GS, such as diarrhea and neutropenia [47,48]. Several antiviral medications, including atazanavir, ribavirin, and pazopanib, can induce hyperbilirubinemia in patients with GS by inhibiting bilirubin-uridine diphosphate glucuronosyltransferase (UGT) activity [49-51]. Additionally, GS is associated with an increased risk of cholelithiasis in adults and children [52,53].
This study had some limitations. First, the number of participants was small and needs to be increased to determine the statistical significance of the clinical relationships among groups with various UGT1A1 genetic variations. Second, although no significant demographic differences were found between the groups that underwent mutation analysis and those that did not, there may still be selection bias in the recruitment process. Finally, as a single-center study that cannot guarantee reproducibility and generalizability, multicenter research with more cases is warranted to better understand the prevalence of UGT1A1 mutations and their clinical relationships.
In conclusion, this study showed that infants with prolonged unconjugated hyperbilirubinemia have a high likelihood of UGT1A1 mutations if the possibility of specific liver or thyroid disorders is excluded. Therefore, UGT1A1 mutation analysis may be beneficial and should be considered an evaluation tool for identifying the underlying cause of prolonged jaundice in infants. It will also be helpful to ease the patient’s parents’ anxiety and provide information as to why their child had jaundice for a long time.
 XML Download
XML Download