

 PDF
PDF Citation
Citation Print
Print



Introduction
Gene editing technology has evolved from the 1st generation zinc finger nucleases and 2nd generation transcription activator-like effector nucleases (TALEN) to the 3rd generation clustered regularly interspaced short palindromic repeats (CRISPR) system. Among them, CRISPR is a genome editing technology derived from bacterial adaptive immunity. Cas protein is programmed to cleave the target DNA following single guide RNA (sgRNA) (1). Target sequences induce double-strand breaks (DSBs) in the DNA and introduce insertion and deletion (indel) mutations that enable cell line or animal modeling via the non-homologous end joining (NHEJ) pathway (2, 3). The donor DNA is processed together, a specific sequence can be knocked in through homology-directed repair (HDR), another mechanism of DNA mismatch repair (4, 5). Furthermore, the expression level of the targeted gene can be regulated by binding a transcriptional repressor or activator to dead Cas (dCas), a Cas protein incapable of DNA cleavage (6, 7). Gaudelli et al. (8) and Komor et al. (9) proposed a base editor that could replace a specific base pair (C∙G to T∙A or A∙T to G∙C) without causing a DSB or externally adding a donor DNA. In 2019, this group also introduced a prime editor that can induce various mutations by inserting a desired sequence into the target position using reverse transcriptase (RT) (10). The development of such gene editing technology has made it possible to introduce and correct various mutations in the DNA sequence of organisms.
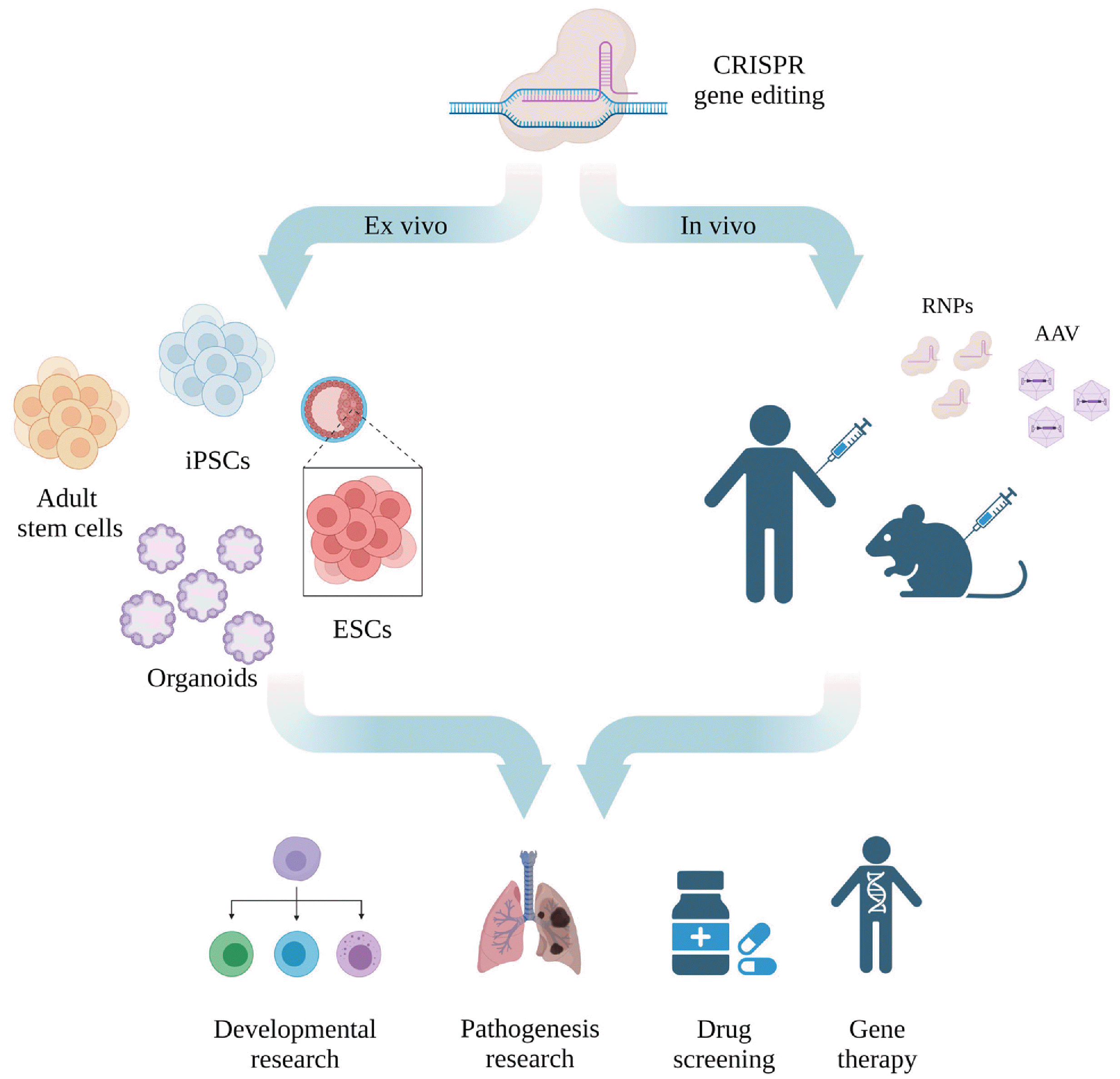
Stem cells are the earliest cell type in the cell lineage that can continuously proliferate and differentiate into various cell types (11). Embryonic stem cells (ESCs) with pluripotency are isolated from the inner cell mass of the blastocyst and are capable of self-renewal and differentiation into other specific cell lineages (11). Even in adults, the stem cells exist in different forms, such as intestinal stem cells (ISCs) that can differentiate into mature cell types necessary for normal intestinal functions and hematopoietic stem cells (HSCs) that can produce blood and immune-related cells (12, 13). However, since adult stem cells can differentiate into the cells of a specific lineage, their differentiation capacity is limited. To solve this problem, in 2006, Takahashi and Yamanaka (14) established a mechanism for differentiating mouse fibroblasts into induced pluripotent stem cells (iPSCs) by regulating the expression of Oct3/4, Sox2, c-Myc, and Klf4 genes. Since patient-specific iPSC production is achievable through this approach, disease modeling has become possible in recent years. This system is expected to be used in patient-specific drug screening or transplantation for cell therapy without triggering an immune response (15). This review describes CRISPR-based gene editing technology used in stem cells and its limitations and potential to be exploited for disease treatment (Fig. 1).
Mechanisms of various CRISPR systems
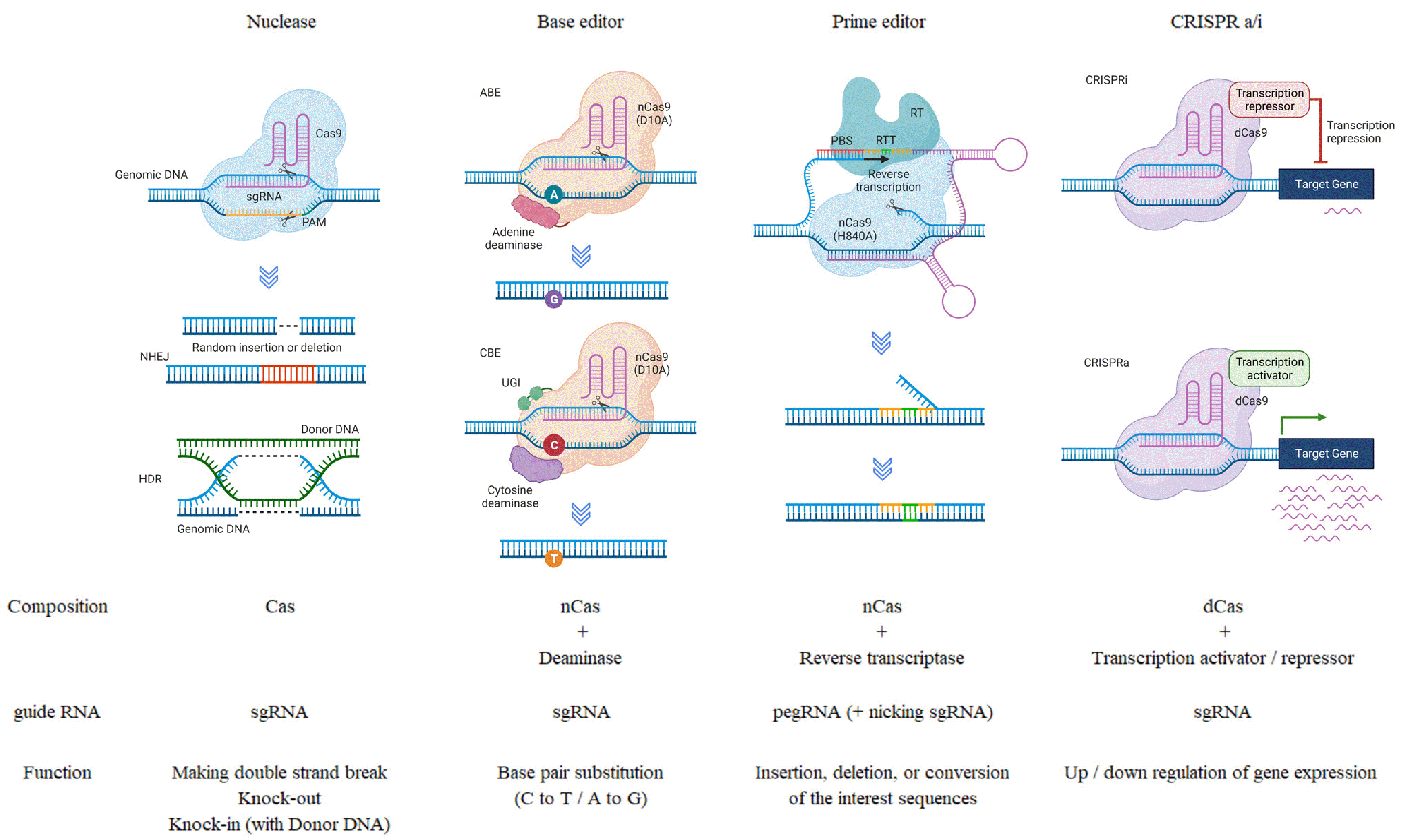
In 1987, CRISPR was first identified in Escherichia coli with the discovery of short tandem repeats interfering with the sequence (16). Since then, through rapid research, development and evolution over the past decade, CRISPR-based genome editing was finally demonstrated in human cells in 2013 (17-19). Here, we provide information on advanced genome editing technologies based on CRISPR (Fig. 2).
Cas (CRISPR-associated) proteins
The CRISPR/Cas system was first identified in 2012 as an adaptive immune system that responds to virus invasion in bacteria by the research team of Jinek et al. (1). The CRISPR/Cas system consists of two components: a Cas nuclease and a sgRNA of 18∼20 bp that guides Cas to the desired DNA location. sgRNA consists of a tracrRNA binding sequence for Cas binding and crRNA complementary to the target sequence (20). SpCas9 derived from Streptococcus pyogenes, a representative Cas nuclease, is widely used for its convenience and high efficiency. In addition, Cas12a (Cpf1) recognizes crRNA-induced T-rich PAM and induces sticky-ended DSB, while Cas13 (C2C2) can edit single-stranded RNA targets (21-23).
The Cas9/sgRNA complex binds to the target sequence and the DNA-RNA hybridization by the sgRNA generates an R loop (24). After forming the R loop, the target and non-target strands are cleaved by the HNH and RuvC domain of SpCas9, respectively, causing DSB (25). DSB occurrence in the cell initiates various DNA repair processes. NHEJ mainly occurs in mammalian cells, which induces the insertion or deletion of random sequences to generate mutations causing frameshift with a probability of 2 out of 3 (2, 3). Cells also repair DSBs by copying and importing intact alleles via the HDR pathway for correct DNA repair (2, 3). HDR is induced at the target site by simultaneously processing the CRISPR system and donor DNA, having the desired nucleotide sequence enabling accurate knock-in. Alternatively, if a short micro-homologous sequence (5∼25 nucleotides) is there on each strand of the DSB-generating sequence, deletions or insertions of various sizes in the target sequence can be achieved through the microhomology-mediated end joining repair process (26). Through various DNA repair processes, this system can easily produce knock-out or knock-in cell lines in target genes. However, it is essential to solve the complications associated with off-target effects and immune response to use CRISPR/Cas system as a therapeutic agent. Of these, the ribonucleoprotein (RNP) complex of CRISPR/Cas is commonly used to reduce off-target effects. RNP, a complex of Cas protein and sgRNA, works rapidly in vivo for gene editing and reduces off-target effects due to its short half-life compared to other systems (27-29). Furthermore, efforts have been made to reduce off-target effects using truncated sgRNAs, engineered Cas variants, or allosterically regulated Cas systems (30). In addition, various anti-CRISPR (Acr) proteins have been identified from phages to inhibit the CRISPR system and are used to reduce the immune response (30-33). They block the normal role of Cas protein by interfering with Cas protein and crRNA binding, inhibiting DNA binding, or losing its ability of DNA cleavage (34-36).
As such, the CRISPR/Cas system can be applied to a wide range of research fields because it is easy to edit the target genome. In this review, we introduce various CRISPR/Cas systems and provide information on how they have been applied to stem cell research.
Base editor
Although CRISPR/Cas has made many advances in gene editing, it can nevertheless result in unexpected indels, translocations, and chromosomal rearrangements caused by DSBs in non-dividing cells (37-41). A new alternative genome editing tool, the base editor system, was developed to overcome these limitations (42).
The base editor systems, including cytosine base editor (CBE) and adenine base editor (ABE), were introduced by Komor et al. (9) as a CRISPR system that was capable of allowing substitution at the single nucleotide level without inducing DSB. CBE comprises deaminase and nickase (nCas)9 (D10A) and can introduce nucleotide-level mutations from C∙G to T∙A in the target sequence (9, 43, 44). Cytosine deaminase replaces C with U in the base editing window, and nCas9, with the D10A mutation, nicks the non-target strand. Further, the complementary G is replaced with an A through cellular mismatch repair. Finally, during DNA replication or repair, U is replaced by T. Uracil glycosylase inhibitor was added to the CBE system to prevent the conversion of U to C by innate uracil DNA-glycosylase (9). CBE induces a C-to-T mutation in an editing window located approximately 4 to 8 bases from the distal end of the PAM in the 20 bp protospacer. CBE has reported to be used with various types of deaminases, such as APOBEC1, APOBEC3A, CDA1, and FERNY (42). These systems are used as base editing tools with improved functions to change the target window and increase efficiency.
The ABE system has the same base editing window as CBE and can introduce A∙T to G∙C mutations. The popularity of the ABE system has increased as it can treat nearly 50% of all human pathogenic point mutations (8, 45, 46). It comprises an engineered version of adenine deaminase (TadA) and nCas9 (D10A). In this system, adenine deaminase replaces A with I, which is changed to G during DNA repair or replication (8).
Various attempts have been made using base editing systems to broaden the primary editing window or narrow the editing window to the single nucleotide level within the target sequences while increasing editing efficiency (43). ABE8e with advanced TadAmax was shown to minimize the size of ABE with increased efficiency, while ABE9 (or NG-ABE9e: This system recognizes NG PAM sequences instead of NGG PAM sequences) changes one target A in editing windows (47-50). In addition, several studies have been conducted using cytosine and adenine deaminase in one base editor (SPACE, Target-ACEmax, and A&C-BEmax) (51-53). Recently, CGBE has been reported to have the capability of base substitution from C∙G to G∙C (54, 55). Also, using different PAM sequences of various Cas orthologous such as SpCas9 engineering version (NG and NGA), SaCas9 [NNGRR(T)], and Cpf1 (TTTV) was shown to extend the target sites (21, 56, 57).
About 50% of known human pathogenic mutation variants are point mutations (58). With the development of base editing technology, point mutations can be easily and efficiently introduced at the nucleotide level, which was difficult with the existing CRISPR gene editing method due to low knock-in efficiency.
Prime editor
Prime editor was reported by Anzalone et al. (10) in 2019, which has the capability to induce small insertion/deletion mutations and all nucleotide conversions. Prime editor comprises nCas9 carrying H840A variant and RT and works with pegRNA containing a spacer, primer-binding site (PBS), and RT template. When the spacer is combined with the target DNA sequence resulting in a nick on the non-target strand, PBS binds to the 3’ of the non-target strand, and RT synthesizes the complementary sequence along the RT template containing the desired mutant sequence. A 3’ edited DNA flap is created as the pegRNA is shed, which competes with the 5’ non-edited DNA flap. In addition, the prime editor uses nicking sgRNA to induce cleavage in the unedited DNA strand to increase DNA editing efficiency.
The prime editor is advantageous since it can introduce various mutations regardless of the type of editing since the RT template is copied and imported as it is (10). Recently, it was found that the efficiency of the prime editor could be increased by grafting chromatin-modulating peptide to facilitate access to target DNA sequences. Further, two prime editors can be used to induce large-scale deletions or chromosomal rearrangements (59, 60). Moreover, a new improved version of PEmax using Cas9 (R221K and N394K) variants, codon-optimized RT and mutation of the 34-aa linker with a bipartite SV40 NLS has been introduced. Additionally, the dominant negative MMR protein (MLH1dn) was transiently co-expressed with PEmax in cells to increase editing efficiency compared to the existing prime editor (61). Nelson et al. (62) improved prime editing efficiency using engineered pegRNA (epegRNA) with structured RNA motifs from tevopreQ1 or Mpknot for preventing degeneration and enhancing the stability of pegRNA. In conclusion, the development of the prime editor made it possible to apply more diverse genome editing beyond indel and limited base editing, which shows the scalability of the scope of use of gene editing.
CRISPER activation/CRISPR interference
Gilbert et al. (63) and Qi et al. (64) revealed that nuclease-null Cas9 (dCas9) does not cause DBS and binds to the promoter region in front of the transcription starting site, preventing RNA polymerase access and inhibits transcri-ption. Furthermore, a system was developed with the capability of more powerful transcriptional repression by including transcriptional repressors such as krüppel-associated box (KRAB), WRPW motif, or chromo shadow domain in the 3’ region of dCas9 (63, 65, 66). Thus, we can study gene functions and pathways with a knock-down strategy rather than permanent gene knock-out using the CRISPR interference (CRISPRi) system.
CRISPER activation (CRISPRa) increases target gene expression by conjugating various transcriptional activators to dCas9 that has lost the enzymatic activity. So far, dCas9-VP64 and various second-generation CRISPRa have been introduced to enhance gene overexpression activity (7, 63). In another report, the synergistic activation mediator (SAM) containing VP64 at the 5’ side of dCas and sgRNA with MS2 hairpin structure were used to induce the formation of four MCP-p65-HSF1 complexes at the target sequence, resulting in the strong transcriptional activity (67). Chavez et al. (68) introduced a method using a tripartite activator VP64-p65-Rta (VPR). Further, Tanenbaum et al. (69) developed Suntag, multiple protein-tagging systems using scFv with VP64.
Stem cell research applied with the CRISPR system
Stem cells have self-renewal abilities, and specific types of cells like mesenchymal stromal cells have the capacity to release various factors, including VEGF, FGF, HGF, PGF, MCP-1, SDF-1, and Ang-1. In this respect, they can be used as promising materials in pathological mechanism, treatment, and regenerative medicine research by applying gene editing technology. Here, we summarize the studies that have employed gene-editing tools to stem cells for treating specific diseases (Table 1) (70-73).
Human pluripotent stem cells
ESCs are pluripotent stem cells with self-renewal capacity and the ability to differentiate into specific cell in the body. Here, we review several research cases exhibiting basic and treatment research by utilizing various gene editing technologies (74, 75) in ESCs.
The development of gene editing systems could greatly facilitate site-specific mutagenesis of human embryonic stem cells (hESCs), including introduction and modification of patient-specific mutations for disease modeling. Zhu et al. (76) demonstrated the strategy for knock-in GFP or RFP in a target gene without drug selection for both active and silent genes in hESCs using TALEN and CRISPRi. Zhou et al. (77) reported the generation of a lineage-specific hESC reporter cell line using the CRISPR/Cas9-system-mediated knock-in method.
Habib et al. (78) reported the development and application of inducible gene editing systems, such as iCas9, iCBE, iABE, and iPE2 to ESC. The possible therapeutic application was also demonstrated by repairing the E342K mutation in the SERPINA1 gene that causes α 1-antitrypsin (A1AT) deficiency in ESCs using the prime and base editors (78).
Kearns et al. (79) has constructed a lentivirus-based doxycycline-inducible CRISPRa/i system in ESCs (Table 2). Through this approach, dCas9-VP64 was used to induce the overexpression of SOX17, and dCas9-KRAB was used to suppress OCT4 to reveal the possibility of differentiation into other cell lineages (Table 2) (79).
Gene editing research in ESCs can be usefully applied to pathogenesis research and therapeutic development by elucidating the function of genes in the process of developing into each cell type and modeling genetic diseases.
Because iPSCs reprogram somatic cells from adult human, they can be used for patient-specific treatment or individual drug screening. In addition, iPSCs provide a way to achieve pluripotency without using embryos.
Park et al. (80) demonstrated mutation-correcting endothelial cell transplantation for the treatment of hemophilia for the first time. iPSCs derived from hemophilia patients with coagulation factor VIII gene mutations were restored to normalcy by CRISPR/Cas9 and subsequently differentiated into endothelial cells. Further, endothelial cells were transplanted into a hemophilia mouse model to examine its therapeutic effects.
Miki et al. (81) also demonstrated mucopolysaccharidosis-targeted gene editing in mouse iPSCs. Neomycin resistance was abolished by delivery of the CRIPSR/cas9 system along with donor DNA into iPSCs to remove a neomycin cassette in exon 6 of the defective α-L-iduronidase terminus.
Rett syndrome (RTT) is induced by mutations in methyl-CpG binding protein 2 (MECP2) and results in slow brain growth and intellectual disability (82). For RTT research, Le et al. (83) introduced the R270X mutation in the MECP2 gene in iPSCs with 20%∼30% efficiency by applying HDR based on the CRISPR system.
The Cys112Arg variant of apolipoprotein E4 (APOE4), a genetic risk factor for sporadic Alzheimer’s disease (sAD), was introduced into iPSCs using a CRISPR/Cas9-based knock-in method to investigate cell type-specific functions of APOE4 concerning AD pathology. The differentiated neuron cells with the APOE4 Cys112Arg variant, such as neurons, astrocytes, and microglia, resulted in extensive gene expression alterations and multiple cellular phenotypes potentially associated with AD pathogenesis (84).
Human urinary cells can be collected non-invasively and directly reprogrammed in iPSCs (85). Yang et al. (85) demonstrated CRISPR/Cas9-based HDR in urine-derived iPSCs from β-thalassemia patients with β-41/42 mutations to treat the mutation by inducing a TCTT deletion between the 41st and 42nd amino acids of HBB gene. This study is a meaningful result showing that the CRISPR system can be provided as a strategy for personalized treatment of β-thalassemia.
AGXT mutations cause excessive accumulation of oxalate in the liver and transport oxalate to the kidneys for-ming insoluble calcium oxalate, which results in primary hyperoxaluria type 1 (PH1) (86). Estève et al. (87) generated functionally corrected hepatocyte-like cells by knock-in the AGXT minigene fragment into AAVS1 locus from iPSCs of PH1 patients.
Mykkänen et al. (88), Vuorio et al. (89), Jalil et al. (90) used the base editing system for patient-derived iPSCs to rescue NOTCH3 (R133C) mutation, a dominant mutation in cerebral autosomal dominant arteriopathy with subcor-tical infarcts and leukoencephalopathy (CADASIL), and LDLR (R595Q) mutation, which causes familial hyper-cholesterolemia.
Chang et al. (91) succeeded in correcting the G2019S mutation in the LRRK2 gene, which is widely known as a mutation in Parkinson’s disease. They also found that ABE had higher on-target editing efficiency, lower off-targets, and indels than CRISPR/Cas9-based HDR in iPSCs.
Chemello et al. (92) used the CBE to replace T with C in the splicing donor sequence of Exon 50 in DMD to treat Duchenne muscular dystrophy (DMD) in iPSCs carrying the ΔExon 51 mutation in the DMD gene. Alternatively, two nucleotides were inserted into DMD Exon 52 of iPSCs using the prime editor. The edited cells were differentia-ted from cardiomyocyte, and the expression level of dystrophin was restored (92).
Spinal muscular atrophy (SMA) is an autosomal recessive motor neuromuscular disorder that weakens muscles and foils normal movement. Most patients with SMA are characterized by exon 7 skipping in the SMN2 transcript, producing unstable truncated proteins. Zhou et al. (93) inser-ted full-length SMN into SMA patient-derived iPSCs by inducing a targeted 9bp-deletion in the intronic splicing silencer-N1 of SMN2 with a prime editor. In the motor neurons derived from rescued iPSCs, apoptosis was reduced by restoring SMN protein. This result suggests that SMA disease caused by skipping exon 7 of the SMN2 gene can also be treated using the prime editor.
To induce neuronal differentiation, the expression of NEUROG2 and NEUROD1 was activated in iPSCs using dCas9-VP64 and dCas9-VPR (Table 2) (68). This study demonstrated the possibility of differentiating iPSCs to neurons through CRISPR-based gene expression activation, and in particular, showed that VPR system was more efficient than VP64 system.
Neural stem cells (NSCs) derived from iPSCs contain microRNA called the miR-199a/214 cluster, a negative regulator of hypoxia-induced cell migration. Luo et al. (94) suppressed the expression of micro RNA in iPSCs-derived NSCs using CRISPRi. This study has contributed to increasing the potential of NSCs for treating neurodegenerative diseases.
Adult stem cells
Adult stem cells have multipotency and can differentiate into several types of cells depending on their origin, such as HSCs, mesenchymal stem cells (MSCs), and NSCs (95). Because these stem cells are derived from adult tissue, they are suitable for targeting organ- or tissue-specific diseases.
Using the Cas nuclease, DNA cleavage can be easily induced at a desired location. The resulting random indel can change the amino acid sequence or induce a frameshift mutation to construct a knock-out model stem cell line. To understand the regulatory mechanism in mouse and human-derived hematopoietic stem and progenitor cells (HSPCs), functional studies have been reported by knocking out EED, SUZ12, and DNMT3A genes using Cas9/sgRNA RNPs (96, 97). AIDS, caused by HIV-1 infection, is generally treated using antiretroviral therapy (ART) (98). However, an effective treatment alternative is needed since ART requires long-term treatment, is expensive, and has various side effects. A few attempts have been made to induce resistance to infection by producing mutations in CCR5, a receptor through which HIV-1 enters. CD34+ HSPCs were treated and transplanted into humanized mice using the CRISPR system, and HIV-1 resistance was also confirmed in second transplantation (99). In addition, the lentiviral vector-mediated system was used to successfully increase resistance against HIV-1 infection in CD4+ T cells and HSPCs through the SaCas9, an easily deliverable small Cas ortholog (99, 100).
Long QT syndrome affects heart repolarization and increases QT length, resulting in abnormal heartbeats (101). Qi et al. (102) modeled an all-in-one episomal encoding base editors, such as epi-ABEmax, epi-ABEmax-NG, epi-AncBE4max, and epi-AncBE4max-NG in HSC to induce mutations in the representative LQT-related genes, KCNQ1 (L114P, R190Q), KCNH2 (Y616C, Y475C), or SCN5A (E1784K). In addition, using a base editor to generate stem cell lines harboring SCN5A (R1879W) mutant associated with Brugada syndrome showed that heart disease modeling is possible in stem cells (102, 103).
Diseases such as sickle cell disease (SCD) and β-tha-lassemia, caused by single nucleotide mutation, can be potential targets for stem cell gene editing. To treat SCD, Cas9 RNPs, and ssDNA donor nucleotides were used to edit a pathogenic point mutation located at the β-globin gene (HBB) of six different patient-derived CD34+ HSPCs (104). Also, Daniel Bauer’s group introduced BCL11A erythroid enhancer +58 C>T and HBB promoter −28 C>T mutations in SCD and β-thalassemia patient-derived HSCs using A3A (N57Q)-BE3 RNPs to ameliorate globin chain imbalance and red blood cell sickling with reduction of bystander mutation near the target nucleotide C (105). If HBB has an E6V (c.17A>T) mutation in SCD, ABE cannot reverse it to wild type. However, T∙A can be changed to C∙G, leading to non-pathogenic E6A, Hb-Ma-kassar (HBBG) (106). To apply these mutations, Chu et al. (107) tried using inlaid base editors to control and increase the base editing window efficiency. In addition, Chu et al. (107), Newby et al. (108), and Miller et al. (109) used a base editor-NRCH capable of non-G PAM (NRCH motif, H=A, C, T) targeting with increased targeting flexibility to edit human and mouse HSCs (A to G conversion, Val to Ala) and transplant them into mice to identify the therapeutic effect (108).
Mutations in SGCA (c.157G>A) cause limb-girdle muscular dystrophies, which weakens the shoulder and pelvic girdle muscles (110). In the study of Escobar et al. (110), the SGCA mutation (c.157G>A) that can induce exon ski-pping was corrected to normal by ABE treatment in muscle stem cells (MuSCs). This result showed that the expre-ssion of SGNA, which encodes α-sarcoglycan, was increa-sed in corrected MuSCs.
Since the use of MSCs avoids ethical issues and tumorigenesis, it is applicable for developing clinical applications including cell therapy and transplantation. The Furuhata et al. (111) induced differentiation from MSCs to white adipocyte-like cells using the target gene transcriptional activation system dCas9-SAM to induce programming of MSCs for therapeutic applications. This approach showed that overexpression of PPARG and CEBPA in MSCs could induce them to become beige adipocyte-like cells, suggesting that this system may also be applied to cell therapy (Table 2).
Stem cell-derived organoids
A few studies have shown gene editing in stem cell-derived organoids. Since stem cell-based organoids have the characteristics of each organ, they are advantageous for detecting diseases caused by genetic mutations. Schene et al. (112) used liver and intestinal organoids to mimic liver cancer growth by inducing a 6 bp deletion in CTNNB1. DGAT1 and ATP7B mutations in patients with congenital diarrhea and Wilson’s disease were restored in ISC organoids using prime editor (112).
Moreover, HDR, a more sophisticated DNA repair method in the CRISPR system, makes it possible to insert desired genes or mutations using donor DNA into precise locations. Schwank et al. (113) corrected ISC organoids derived from cystic fibrosis (CF) patients induced by the F508del mutation in CFTR to normal by a CRISPR system-mediated HDR technology. This group also performed cancer development modeling by applying the R175H, R249S, and Y220C mutations of TP53 using hepatocytes and colonic organoids via prime editing (114). This study demonstrated that the F580del and R785X mutations in CFTR that induce CF could be repaired using a prime editor in patient-derived gut organoids (114). Collectively, these research reports suggest that it is possible to efficiently introduce and correct mutations in 2D cells and 3D culturable organoids using various gene editing tools, which can be applied to various basic and therapeutic studies.
Future perspective
Stem cells are pluripotent and have significant advantages in cell differentiation and pathogenic studies. With the availability of single-cell analysis, lineage tracing has also become feasible. Recent advances in ex vivo and in vivo genome editing technologies have emerged as new therapeutic approaches with great potential for correcting genetic mutations in targeted stem cells. In this study, we summarized research trends about various CRISPR systems applied to stem cells. Not only gene knock-out/-in but also various types of editing, such as one nucleotide substitution and large insertion/deletion, are achievable through the CRISPR system. However, methods for precise editing as well as efficient and safe delivery without unwanted mutations in vitro and in vivo must still be developed before genome editing can be approved as a therapeutic tool. Moreover, there is still the off-target problem of editing sequences similar to the target, which needs to be addressed in the future for more sophisticated stem cell editing.
Recently, DNA DSBs introduced using Cas/sgRNA have been shown to cause deletions, inversions, and clone creations extending over many kilobases (115). Although difficulties in delivery and off-targets have been raised as limitations, CRISPR is still an irreplaceable and valuable tool for basic or clinical research. Furthermore, miniature Cas orthologs such as Cas12f have been discovered, enabling easier delivery (116). If various CRISPR systems are adequately utilized, it will significantly help stem cell research, such as lineage differentiation studies, disease modeling, and ex vivo treatment.
 XML Download
XML Download