

 PDF
PDF Citation
Citation Print
Print



Vascular Endothelial Cell Development and Diversity
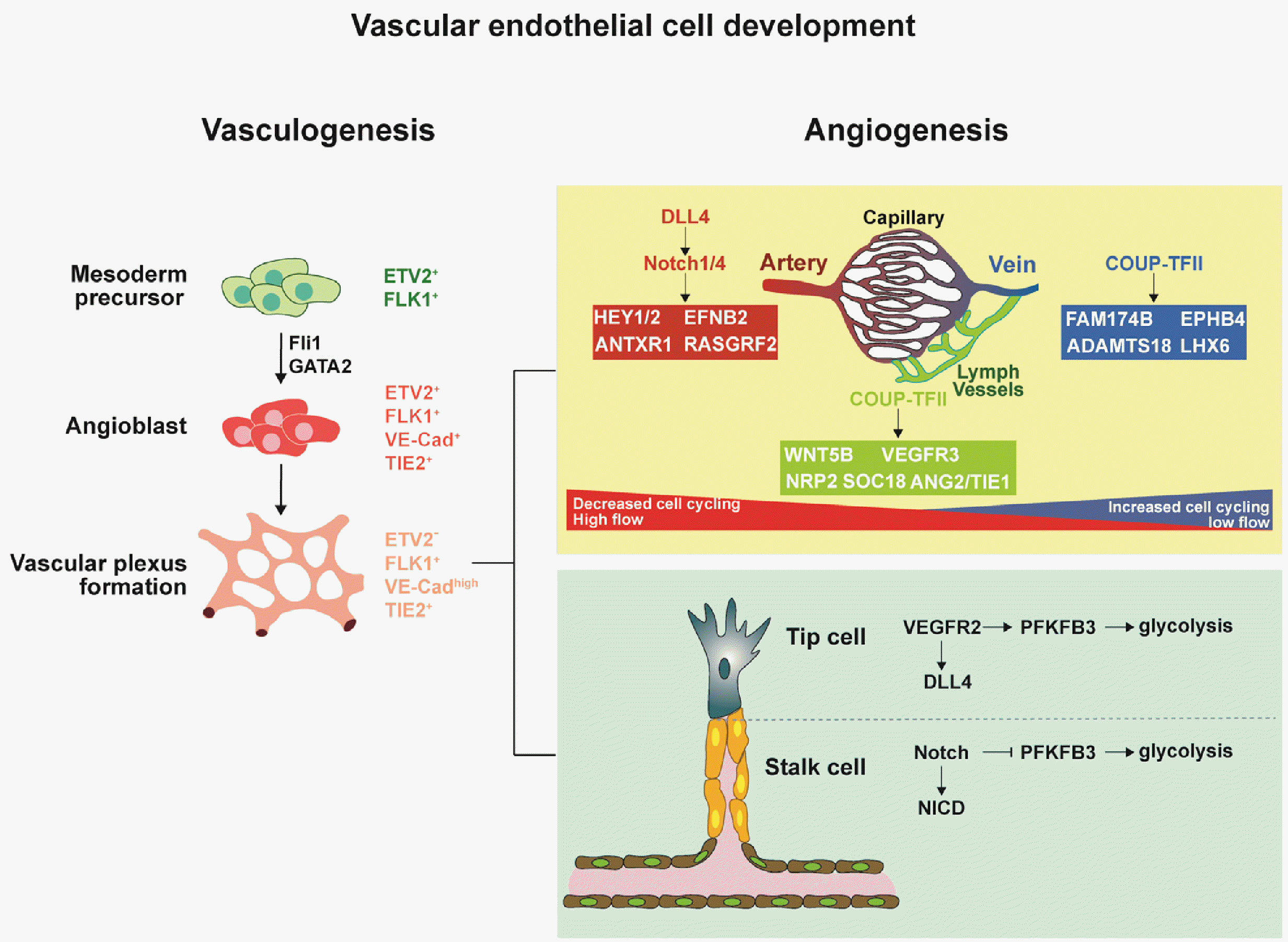
Vasculature is a closed hierarchical network of arteries, veins, and interconnecting capillaries that specifies in different tissues. Vascular development initiates during embryogenesis through the intricate coordination of critical regulators that drive vessel formation, stabilization, branching, and pruning. Two major processes are involved in vascular development, vasculogenesis and angiogenesis. During vasculogenesis, mesodermal precursors (ETV2+, FLK1+) differentiate into angioblasts, forming an early primitive vascular plexus before specifying into the dorsal aorta, cardinal vein and subsequently arteriovenous blood vessels (Fig. 1, left panel) (1). New vessels further develop via angiogenesis and vascular remodeling into a hierarchical vascular network. Each crucial step of differentiation and remodeling necessitates the integration of both chemical and mechanical signals, which trigger morphogenic behaviors (Fig. 1, right upper panel).
One of the critical signaling pathways driving initial vascular development and angiogenesis is the vascular endothelial growth factor (VEGF) signaling pathway. Maintaining a balanced level of VEGF is particularly crucial for establishing a proper vascular network. Studies conducted on rodents have demonstrated that the absence of a single VEGF allele can lead to abnormal vessel development and lethality in mouse embryos (2) whereas overexpression of VEGF results in embryonic lethality (3) in mice. VEGF comprises various classes with different homodimeric ligands, with VEGF-A playing the most critical role in promoting angiogenesis and developing nascent vessels. VEGF-A isoforms exert their activity by interacting with kinase tyrosine receptors on endothelial cells (ECs), namely VEGF receptor 1 and VEGF receptor 2 (VEGFR2). VEGFR2, expressed on vascular endothelium, is the most predominant receptor involved in VEGF signaling (3). Upon binding of VEGF-A, receptor dimerization occurs, leading to the phosphorylation of tyrosine residues and initiation of signaling pathways that regulate angiogenesis, endothelial proliferation, survival, migration, and vascular permeability (4).
Another critical component for vascular development is biomechanical stimuli. Hemodynamic flow and pressure induce crucial signals for maturing and stabilizing the vasculature into hierarchical networks. One critical mechanosensitive signaling pathway involved in this process is Notch, which plays a versatile role throughout blood vessel development. In the early stages of development, Notch is essential for expanding vessels from the vascular plexus (5). Deletion of Notch1 in transgenic mice before E9.5 results in embryonic lethality, while deletion at the 8-week or later stage leads to the loss of cell-cell junctions and the absence of mechanosensing in arteries (6, 7). Notch signaling is also heavily involved in angiogenic expansion of vascular beds during the formation of tip and stalk cells in angiogenic sprouts. During angiogenic sprouting, two types of cells emerge: tip cells located at the sprout’s leading edge and stalk cells adjacent to the tip cells, which proliferate and form new blood vessels. Angiogenesis is initiated in response to VEGF gradients, which activate the Notch receptor delta-like 4 (DLL4) via VEGFR2 (6). Activation of DLL4 in tip cells leads to their transition from a proliferative state to a migratory state. Subsequently, the interaction between DLL4 and Notch ligand receptor in tip cells activates Notch signaling and triggers the translocation of the Notch intracellular domain, suppressing the tip cell phenotype (Fig. 1, right lower panel) (7). Downstream of VEGFR2 and Notch signaling in tip-stalk cross talk is the PFKFB3 gene, which is responsible for stimulating glycolytic activity in cells. ECs exhibit high glycolytic activity, generating approximately 85% of their ATP through glycolysis. This is consistent with ECs having a smaller mitochondrial volume fraction (5%) than other cell types. Studies have shown that 6-phosphofructo-2-kinase/fructose-2, 6-bisphosphatase (PFKFB3) activity is increased in response to sprout promoting signals such as VEGF, while PFKFB3 activity is significantly reduced in response to sprout limiting signals such as DLL4 (8). Silencing of PFKFB3 leads to immobile filopodia, quiescent tip cells, and decreased proliferation in stalk cells suggesting that PFKFB3 is important for regulating glycolysis and cytoskeletal organization, which are crucial for promoting functional angiogenesis (9).
Following vasculogenesis and angiogenesis, the newly formed vessel network undergoes stabilization to establish an intact vasculature with the assistance of mural cells that surround the abluminal lining of ECs (10). The platelet-derived growth factor-B (PDGF-B) – PDGF receptor β (PDGFRβ) pathway and Angiopoietin (ANG)-TIE pathway are two crucial pathways implicated in vascular stabilization and the maintenance of vascular homeostasis. PDGFRβ is predominantly expressed in mural cells, including vascular smooth muscle cells and pericytes, and is primarily involved in regulating the proliferation and recruitment of these mural cells towards nascent vessels (11). PDGF signaling plays a fundamental role in vessel development, as the deletion of PDGF results in the failure of mural cell recruitment and leads to embryonic lethality. In the presence of VEGF-A signals, ECs enhance the expression of PDGF-B, which interacts with PDGFRβ on mural cells and initiate signaling cascades involved in proliferation and migration, such as phosphoinositide 3-kinase (PI3K), extracellular regulatory kinase 1/2, and mitogen-activated protein kinase (MAPK) (11). Conditional inactivation of PDGF-B secretion by ECs prevents the migration and proliferation of mural cells, suggesting that ECs are the primary source of PDGF-B required for vascular stabilization (12).
The ANG-TIE signaling pathway, crucial for lymphatic vessel development, also significantly stabilizes newly formed vessels and regulates vessel permeability (13). ANG (consisting of growth factors ANG1-ANG4) interacts with TIE2 receptors expressed on ECs. ANG1, a potent TIE2 agonist, promotes enhanced EC survival and barrier function by stabilizing vascular endothelial cadherin (VECAD) junctions. The binding of ANG1 to TIE2 triggers the formation of TIE clusters on the surface of ECs, facilitating cell-cell junctions. Moreover, ANG1 aids in the translocation of TIE receptors towards extracellular matrix (ECM) substrates, promoting enhanced cell-substrate contacts (14). Downstream signaling pathways in ANG1-TIE interaction involve activating PI3K/AKT pathways, inhibiting the transcriptional factor Forkhead box O1 (FOXO1) and its targets, such as ANG2 (15). In contrast to ANG1, ANG2 acts as a weak agonist and antagonist to TIE2 in a context-dependent manner (16). The expression of ANG2 is increased in response to inflammatory signals or VEGF stimulation, which reduces ANG1-TIE2 signaling. This reduction in ANG1-TIE2 signaling, in turn, leads to further upregulation of ANG2 expression through FOXO1 inhibition. ANG2 also induces endothelial apoptosis without VEGF signaling, contributing to vascular rarefaction (17).
The orchestra of VEGF (18), Notch (19) and other signaling pathways such as Hedgehog (20) and Wnt/β-catenin (21) govern the arteriovenous specification during development. VEGF acts as the primary driver of arterial specifications through direct stimulation of DLL4/Notch (18) or indirect stimulation of ETS factors (22). Different VEGF isoforms also preferentially direct vessels towards arteriovenous specification. In mice designed to express single VEGF isoform, VEGF164 was sufficient to direct arterial specification, and VEGF188 was necessary for venular development (23). Arterial fate is also driven by the activation of transcription factors (TFs) Forkhead box (FOX) C1 and FOX C2, which leads to increased expression of DLL4/Notch and downstream signals such as EFNB2 and HEY/HES (Fig. 1, right upper panel) (24). In contrast, venous specification is marked by the expression of COUP transcription factor 2 (COUP-TFII), which is required to suppress the Notch signaling pathway (25). This suggests that vessels are programmed to follow arterial fate during hierarchical vessel formation, and the repression of Notch signaling is important for hierarchical vessel formation. However, studies show that vessels are not locked into its “pre-determined” arteriovenous subtypes but rather respond to hemodynamic forces that alter expression profile towards arterial or venous subtypes (26, 27).
Moreover, cell cycling influences EC fate determination in arteriovenous specification (Fig. 1, right upper panel). The Notch and COUP-TFII signaling pathways play crucial roles in the arterial differentiation of ECs by influencing cell cycling and metabolism rather than directly regulating arterial and venous fate determinants (25). Studies in various model systems, such as the retina and developing heart, have revealed that shear stress-induced Notch signaling leads to cell cycle arrest and the expression of arterial specification genes (28, 29). On the other hand, COUP-TFII inhibits arterial differentiation by activating cell cycle genes. Epigenetic studies have supported the connection between cell cycle genes and COUP-TFII’s inhibitory effects (30, 31). Proliferating ECs exhibit a bias towards venous fate, as indicated by the expression of venous EC genes. Recent research has demonstrated that ECs in the late G1 state respond more to arterial specification signals (32). Additionally, arterial pre-specification has been observed in different vascular beds, involving low proliferative rates, high Notch activity, and VEGF-ERK signaling.
Our body has meticulously mapped out the signaling pathways that govern and sustain vascularization throughout development and homeostasis. A deeper comprehension of the intricate interactions among these signaling pathways in both spatial and temporal contexts offers us opportunities to enhance vasculature construction in vivo, regulate appropriate vascularization in engineered tissues in vitro, and facilitate vascular regeneration in vivo.
Hemogenic Endoderm and Immune Cell Differentiation
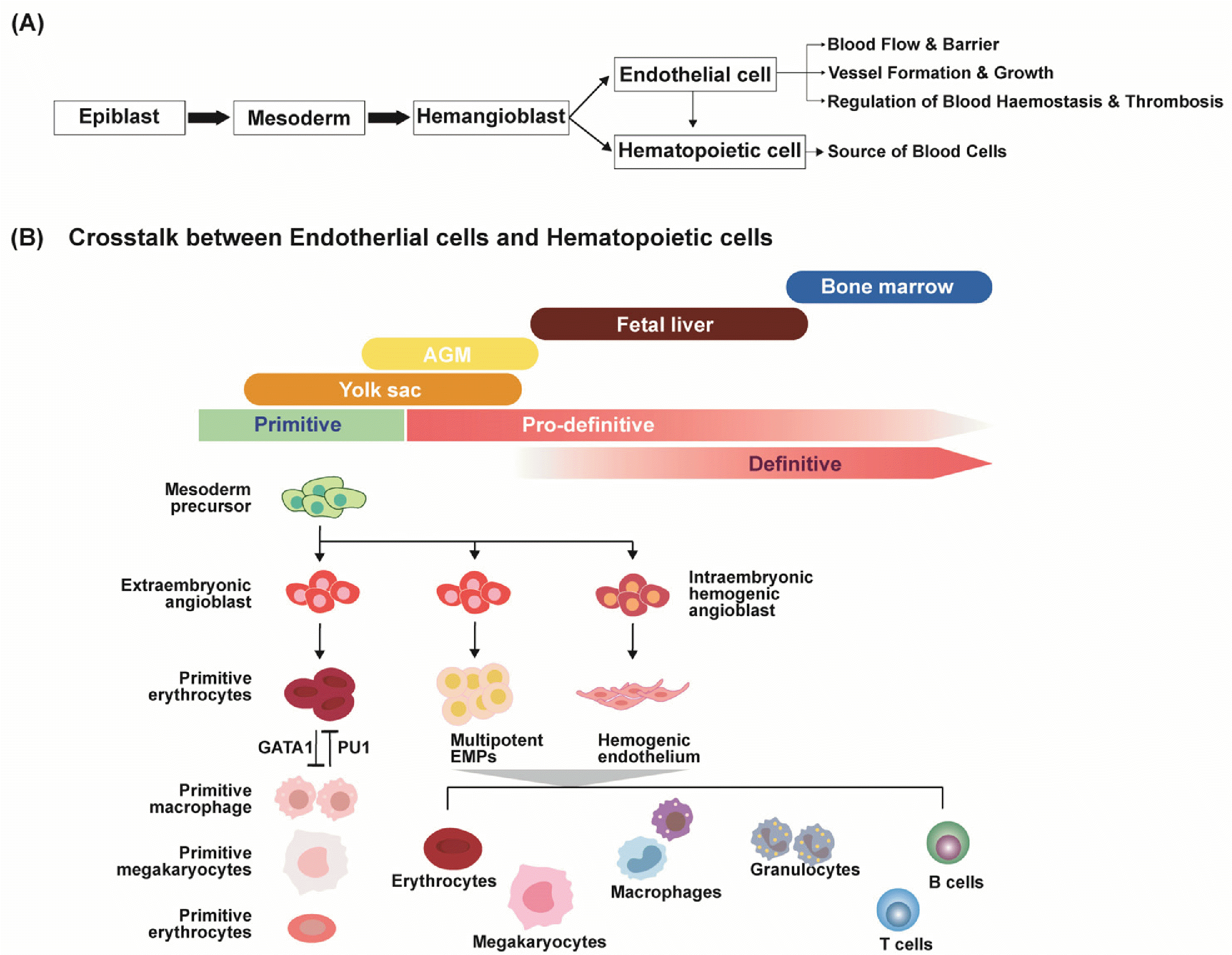
The establishment of the adult blood and immune system relies on the emergence of hematopoietic stem cells (HSCs) and progenitors, which initiate during embryonic development prior to the formation of functional circulation. This process, known as hematopoiesis, occurs in multiple stages (primitive and definitive hematopoiesis) where spatial and temporal developmental waves give rise to distinct populations of hematopoietic cells (Fig. 2) (33). During vertebrate fetal development, ECs and hematopoietic cells exhibit close relationships, sharing the same developmental trajectory as mesodermal progenitors. As the embryo develops, a subset of hemogenic ECs derived from hemogenic angioblasts undergoes endothelial-to-hematopoietic transition (EHT) (34). The transition involves the acquisition of hematopoietic-pecific markers and loss of EC-specific markers, leading to further differentiation into adult HSCs.
Primitive hematopoiesis occurs in two waves (Fig. 2), where the initial wave of hematopoiesis leads to generation of cohorts of transient, tissue-resident hematopoietic cells from the angioblasts of the extraembryonic yolk sac (35). The initial cohort of cells (E7∼E7.5) includes primitive erythrocytes, macrophages and megakaryocytes, which support embryo growth through oxygenation, tissue remo-deling and lymphatic development (36). The cells generated during this phase lack pluripotency and self-renewal capacity and are thus transient. The emergence of these primitive erythroid and myeloid cells is determined by the transcriptional factors GATA binding protein 1 (GATA1) and PU.1. GATA1 and PU.1 provide cross-inhibitory regulation of erythroid versus myeloid fate, where the suppression of GATA1 leads to a reduction in myeloid cells and increased expression of PU1, and vice versa (37).
During the second wave (E8.25) of primitive hematopoiesis, multipotent erythroid and myeloid progenitors (EMPs), a subset of B lymphocytes (B-1a cells) and T lymphocytes emerge from intraembryonic vascular plexus (38, 39). EMPs that arise during this phase differentiate into cerebral glial cells found in adult brain tissues (40) as well as small populations of tissue resident macrophages found in lung and liver (41). Important TFs that regulate EMP formation during this phase are Runx1, Csf1r, and Ckit (40, 42, 43). Lineage tracing studies have shown that controlling such TFs leads to altered levels of macrophage progeny in the adult mice, in the absence of the c-myb gene required for adult HSC developmen (44). B-1a cells, a subclass of B lymphocytes that regulate immunological tone and homeostasis, a subset of para-aortic splanchnopleura-derived T lymphocytes, and RAG1+ multipotent T cells with myeloid potential are also thought to arise from this pro-definitive phase of hematopoiesis (38, 45, 46).
Finally, the third wave of hematopoietic cell generation leads to definitive hematopoiesis, which produces adult-type HSCs responsible for generating the majority of hematopoietic cells in the blood and resident tissues (Fig. 2) (47). HSCs originate from a small subset of embryonic ECs known as hemogenic ECs, first identified in the aorta-gonad-mesonephros region at E10.5 (48). These hemogenic ECs undergo a downregulation of EC-specific molecular signatures and activate transcriptional programs necessary for the emergence of hematopoietic progenitors. Several studies in vertebrate embryos suggest that functional HSCs are initially found in the wall of the dorsal aorta in the aorta–gonad–mesonephros (AGM) region in mouse embryos (48-50). Furthermore, these hemogenic ECs differentiate into HSCs exclusively within the AGM region, suggesting that HSC fate is determined by extrinsic signals from the arterial vascular niche (51). Key TFs that regulate EHT include Runx1, GATA2, Gfi1, and Gfi1b (52-54). In zebrafish, a subset of Flk1+ hemogenic ECs acquire the definitive hematopoietic marker RUNX1 and subsequently develop into HSCs (55). Notably, Notch1-induced GATA2 expression serves as an essential temporal signature of HSC development (56). Prolonged expression of GATA2 leads to non-functional hematopoietic progenitors and impaired HSC development (57). Overall, it is understood that ECs lining the wall of the dorsal aorta with the hemogenic potential must downregulate EC-specific markers and upregulate hematopoietic markers for successful EHT. Following their development in the AGM region, maintaining their stemness within this microenvironment (58).
Molecular Signatures of Tissue-Specific ECs
ECs are among the earliest cells to develop during vascular development, and their formation occurs uniformly in the blood islands of the extra embryonic yolk sac. As ECs vascularize each organ during development, they acquire heterogenous organ-specific properties, including phenotypic, functional, and angiocrine characteristics that match the diverse metabolic and structural demands of each organ. For example, liver sinusoidal endothelial cells (LSECs) that line the liver adjacent to hepatocytes exhibit a high degree of fenestrations to facilitate the transport of macromolecules between the liver and the blood stream (59). In contrast, brain capillary ECs are characterized by an increased presence of tight junction proteins such as claudins and occludins, as well as drug efflux transporters like p-glycoproteins and multi-drug resistance-associated proteins (60). These features contribute to tightening transcellular gaps and restricts the transport of molecules from the bloodsteram to the brain parenchyma. In various tissues, ECs are also known to arise from tissue-resident progenitors, thus possess tissue-specific regenerative capacity and exhibiting distinct responses to local tissue injury (61).
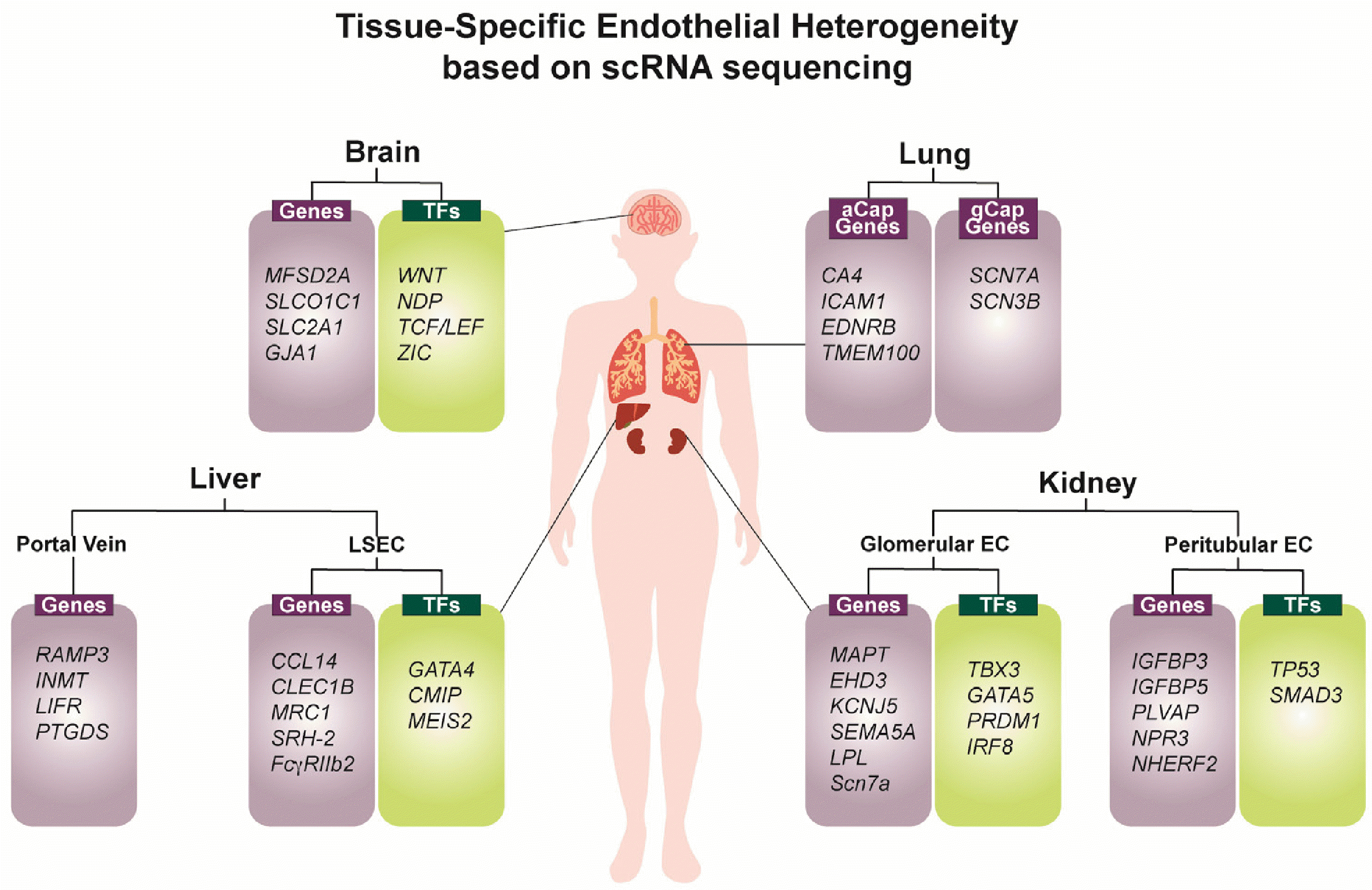
Recently, the development of multimodal omics tools has allowed for transcriptional and epigenetic insights into the heterogeneity of ECs across organs (Fig. 3). Organism-wide investigations using single-cell RNA sequencing (scRNA-seq), notably the Tabula Muris dataset, which provides single-cell transcriptomic profiles of ECs from 12 major organs, has revealed both similarities and disparities in the transcriptomes of ECs among these organs (62-64). Interestingly, studies have shown that lymphatic ECs across different organ systems share a similar transcriptomic profile, while blood vascular ECs exhibit heterogeneity (64). Within the vascular hierarchy, capillary ECs display the most distinctive transcriptomic profile compared to arteries and veins, which share similar transcriptomic signatures across different tissues. Major organs with highly specialized vasculature, such as the brain, liver, kidney and lung, also exhibit distinct EC signatures, demonstrating intra-organ EC heterogeneity (64-66) as well as zone-specific (67-70) gene expression profiles within the tissue. In contrast, tissues such as adipose, skeletal muscle and aorta show overlapping gene clusters. While the developmental trajectories of ECs are similar across the organs, the regulation of WNT and MAPK signaling pathways plays a crucial role in determining tissue-specific gene expression patterns (64). Furthermore, scRNA-seq of ECs across organs has revealed sexual dimo-rphism, particularly in the heart, brain and lung, where the non-sex-linked gene Lars2 is enriched in male murine ECs. This data suggests an additional layer of EC heterogeneity: sex-specific EC transcriptome in major organs (64).
From an epigenetic perspective, the incorporation of Assay for Transposase-Accessible Chromatin sequencing in multiomic studies of ECs has enabled further investigation into the TF motifs that regulate the distinct EC transcriptome (71). Given that ECs share a common developmental pathway during embryonic development, the ETS TF family was found to be globally enriched in all EC sub-types. Further examination of motif enrichment in the commonly expressed TFs in ECs revealed distinct patterns of enrichment corresponding to each EC subtypes (71). In the brain, TFs such as FOX, TCF/LEF and ZIC were identified, all of which are known to regulate WNT signaling, an important pathway involved in blood-brain barrier development and angiogenesis. In the liver, GATA TF families were enriched, which aligns with previous findings implicating the importance of GATA4 in hepatic EC development. In the kidney, Homeobox TF families were strongly enriched, confirming other scRNA-seq findings that show high expression of homeobox transcripts such as Irx3, Hoxa7, Pbx1, and Meis2.
Overall, comparative analysis of scRNA-seq data suggests a strong correlation between parenchymal and EC gene expression. Therefore, it is crucial to understand EC niche interactions, as they play a critical role in maintaining region-specific EC signatures. In fact, primary ECs harvested from tissues and cultured in vitro often lose the transcriptomic and phenotypic signature observed in vivo (65). This suggests that ECs possess their own tissue-specific gene expression, but cell-cell and cell- ECM niche interaction are critical for maintaining their heterogeneity.
Brain
The capillaries of the brain are characterized by tight, continuous, non-fenestrated ECs that tightly regulate the blood-brain barrier, restricting the transport of molecules from the plasma to the central nervous system (CNS) compartment. Generally, the capillary ECs that make up the brain vasculature are VEGFR2+/VECAD+/CD133+ and express low levels of thrombomodulin. scRNA-seq studies have shown that brain ECs express transcripts encoding drug efflux transporters such as Mfsd2a, which prevents transcytosis of various chemicals that would normally pass through the vascular lumen into resident tissues, and lc2a1, a glucose transporter required for efficient glucose transport to meet the high metabolic demand of the brain (69, 71). During development, CNS ECs coincide with the development of neural tissue, suggesting that neuronal signals play a significant role in the acquisition of CNS-specific ECs. Notably, WNT and NORRIN ligands expressed in neurons and glial cells activate EC receptors to maintain blood-brain-barrier and direct CNS angiogenesis. In the subventricular zone (SVZ) where neural stem cell (NSC) populations reside, CNS ECs interact with NSCs to regulate NSC behavior. CX43 expression in ECs in SVZ mediates the proliferation and differentiation of NSCs. In the subgranular zone of the brain, CNS ECs utilize angiocrine factors to regulate NSC proliferation and activation (72).
Lung
The lung is a highly vascularized organ surrounded by a dense network of capillaries that facilitate efficient gas exchange for oxygenation of the blood. Recent scRNA-seq studies have identified two discrete populations of capillary ECs within the alveoli. Interestingly, these two lung EC subtypes did not exhibit a zone-specific signature but were intermingled throughout the gas exchange interface (73, 74). The first subtype, referred to as aerocyte capillary (aCap) ECs, showed enrichment for CAR4, ICAM1, and EDNRB. The CAR4 gene codes for carbonic anhydrase 4, which plays a role in catalyzing the conversion of carbon dioxide into bicarbonate. The enrichment of ICAM1 suggests that aCap ECs are actively involved in antigen regulation and processing. Another scRNA-seq study by Paik et al. (64) also identified a unique cluster of lung capillary ECs enriched for Tmem100, indicating their role as antigen-presenting ECs. This suggests that aCap ECs are actively involved in antigen regulation and processing. The second lung EC subtype found in the alveoli is called general capillary (gCap) ECs. gCap ECs express lung EC-specific markers Scn7a and Scn3b, which encode the sodium channel and likely contribute to the regulation of serum sodium concentration sensing and control of the renin-angiotensin-aldosterone system (73, 74).
Liver
Liver ECs, also known as sinusoidal ECs, play a crucial role in supporting blood transport between the hepatic artery and portal vein, as well as in scavenging molecules for immune clearance. As a result, LSECs are enriched with genes that encode scavenger receptors, including Fcgr2b, Stab2, and Clec4g (75, 76). Similar to the kidney, Liver ECs also exhibit intra-organ specificity with zonation-specific EC signatures. Spatially resolved scRNA-seq studies have reported that portal vein ECs in the liver show increased levels of RAMP3 and INMT (70, 77). LSECs can be further divided into three distinct zones based on their localization relative to the portal vein and central vein. Regions closest to the central vein exhibit markers such as CCL14 and CLEC1B, indicating decreased vascular tone and increased capillarization (70, 78). On the other hand, ECs closer to the portal ECs show an increased level of MGP. However, the most distinct enrichment is observed between LSECs and Portal ECs which are developmentally distinct. Portal vein ECs develop from vitelline veins, while LSECs are capillary ECs that acquire fenestration during embryonic development via TF GATA4 (71).
Kidney
In the kidney, the microvasculature exhibits distinct markers specific to its functional compartments: the renal cortex and medulla. The renal cortex is responsible for the ultrafiltration of blood and contains glomerular capillaries within Bowman’s capsule, which selectively drain amino acids, glucose, urea, and sodium chloride toward the tubules. The renal medulla, on the other hand, regulates urine concentration through the balance of water and solutes. Functionally, the ECs in the kidney also display zone-specific signatures between glomerular ECs, which regulate filtration, and peritubular ECs, which regulate reabsorption and secretion. scRNA-seq studies that aimed to dissect the intra-organ EC heterogeneity revealed that ECs within the renal cortex are enriched with markers such as Igfbp3 and Npr3 and the TF FOXP1 (79, 80). In contrast, ECs within the renal medulla expressed markers In contrast, cells within the renal medulla expressed markers such as IGF-1, CD36, and transcriptional factors like LEF1. Another scRNA-seq study by Barry et al. (67) identified transcriptomic disparities in glomerular capillary (GC) ECs and investigate the potential mechanisms involved in GC specification. This study revealed that GCs specifically expressed transcriptional factors Txb3, Gata5, and Prdm1, which prune the expression profile in GCs and potentiate their differentiation into glomerular capillaries.
Tissue Specific Immunomodulation of Endothelial Cell
ECs, responsible for maintaining vascular integrity and regulating blood flow, also play a critical role in immune responses within the body. They accomplish this through various mechanisms, including antigen presentation, expression of adhesion molecules, and production of cytokines and chemokines. These functions are essential in the context of inflammation, where ECs have a crucial role in modulating immune responses. ECs’ expression of adhesion molecules is particularly important in their interaction with immune cells. For example, the expression of intercellular adhesion molecule-1 (ICAM-1) on ECs enables their interaction with leukocytes, facilitating immune cell recruitment and transendothelial migration (81). In addition, vascular cell adhesion molecule-1 (VCAM-1) expression is upregulated on ECs during inflammation and plays a crucial role in the adhesion and migration of immune cells to inflamed tissues (82). Selectins also play an essential role in the interaction between ECs and leukocytes. ECs express P-selectin, E-selectin, and L-selectin, which mediate the rolling of leukocytes on the endothelial surface and their subsequent adhesion and transendothelial migration (83).
Furthermore, ECs in specific organs produce cytokines and chemokines that tightly regulate immune responses within those organs. These cytokines and chemokines include interleukin-1 (IL-1), interleukin-6 (IL-6), tumor necrosis factor-alpha (TNF-α), and interferon-gamma (IFN-γ) (84). During tissue homeostasis, immune cells patrol in and out of blood vessels providing regulatory surveillance until inflammatory signals activate both immune and ECs. The EC activation and secretion profile can vary in organ systems. For example, TNF-α produced by ECs in the liver during inflammation regulates immune responses by promoting the differentiation and survival of T cells (85). ECs in the lung produce IL-8, a potent chemoattractant for neutrophils, and CCL2, which recruits monocytes to the site of inflam-mation. In the kidney, ECs produce IL-6, a cytokine that promotes the survival and differentiation of T cells, and CXCL12, which recruits immune cells to the site of inflammation. Likewise, in the heart, ECs produce IL-1β, a pro-inflammatory cytokine that promotes the survival and differentiation of T cells, and CCL5, which recruits monocytes and T cells to the site of inflammation. Brain, known to express minimal levels of adhesion molecules, cytokines and chemokines under homeostatic conditions, upregulates expression of adhesion molecules such as E-selectin and enhances the secretion of chemokines such as CXCL1 upon inflammation.
Several studies also provide evidence of several EC subtypes that display antigen-presenting capacity (83). For example, hepatic ECs express high levels of MHC class and co-stimulatory molecules, enabling them to efficiently present antigens to CD4+ and CD8+ T cells (85). This unique ability allows them to modulate the immune response during liver inflammation and injury. Similarly, lung ECs express MHC class and co-stimulatory molecules, enabling them to present antigens to T cells and regulate immune responses within the lung (73). ECs in the kidney, brain, and heart also express MHC class and co-stimulatory molecules, enabling them to present antigens to T cells and regulate immune responses within their respective organs. Additionally, they produce a variety of cytokines and chemokines that play a role in immune regulation within these organs. Furthermore, ECs express adhesion molecules that facilitate their interaction with immune cells and regulate their migration into the organ tissue. Similarly, brain ECs produce IFN-γ, enabling them to present antigens to T cells and regulate immune responses in the CNS by expressing MHC class and co-stimulatory molecules (86).
Understanding the role of organ-specific ECs in regulating immune responses and targeting the immunomodu-latory function holds great promise for developing novel therapeutic strategies for immunological disorders. For example, targeting the interaction between brain ECs and immune cells could represent a promising approach for treating neurological diseases such as multiple sclerosis and Alzheimer’s (87). Given that the blood-brain barrier restricts the entry of drugs into the brain, the development of therapies that specifically target brain ECs can enhance drug delivery to the brain while minimizing off-target effects in other organs. Additionally, autoimmune and metabolic diseases that trigger chronic inflammation necessitate therapeutic strategies that target the immune-EC crosstalk. Overall, gaining insights into the role of organ-specific ECs in immune regulation can pave the way for the development of more targeted and effective therapies for various immunological disorders.
In conclusion, the immunomodulatory function of organ-specific ECs has profound implications for the development of targeted therapies and novel therapeutic strategies for a wide range of diseases. To effectively intervene, further research is needed to unravel the specific mechanisms underlying the interaction between endothelial and immune cells in different organs. This includes identifying the specific adhesion molecules, cytokines and chemokines involved in immune regulation within organ-specific ECs, as well as pinpointing potential targets for intervention. Advancing our understanding in these areas will pave the way for the development of more precise and effective therapeutic interventions for various diseases.
EC and Immuno-Vascular Dysfunction in Disease
Vasculopathies serve as a hallmark of chronic inflammatory diseases including diabetes, atherosclerosis, autoimmune diseases and cancer. Due to the manifestation of chronic inflammation, the crosstalk between immune cells and ECs becomes one of the crucial aspects of disease progression and intervention. Dysregulation of homeostatic immune EC interaction leads to severe and debilitating angiopathies which are accompanied by alterations in signaling pathways that govern EC activation, immune cell recruitment, immune cell tolerance and abnormal vascular growth.
Interconnected mechanisms of diabetes mellitus and atherosclerosis
Diabetes mellitus (DM) is a metabolic disease characterized as a state of systemic hyperglycemia, dyslipidemia and insulin resistance. Type 2 diabetes (T2D), the most common form of diabetes, is characterized by a systemic and chronic inflammatory state. Hyperglycemia in T2D exacerbates the progression of DM by inducing mitochondrial dysfunction and subsequent production of reactive oxygen species (ROS) (88, 89). As such, high level of ROS in the systemic circulation and increased inflammatory responses induce the additional release of pro-inflammatory cytokines and chemokines in the systemic circulation. Therefore, T2D often accompanies severe microvascular complications (e.g., microangiopathy in chronic kidney disease) and macrovascular complications (e.g., atherosclerosis) (90).
Atherosclerosis is a common yet morbid macrovascular complication that arises in most DM patients. Atherosclerosis is characterized by the thinking of the arterial lumen through the deposition of cholesterol plaques and subse-quent obstruction of blood flow. The cholesterol plaques in DM develop as dyslipidemia in DM leads to an increased level of cholesterol and oxidized low-density lipoprotein (ox-LDL) (91). In DM vasculature, hyperglycemia causes glycation of red blood cells, increased adhesion molecules and cytokine release via activated ECs and monocyte recruit-ment. As recruited monocytes differentiate into macrophages, excess lipid uptake in an atherosclerotic environment leads to foam cell formation (92). Interaction of ox-LDL with its lectin-like receptor LOX-1 triggers NADPH oxidase and activates the NF-kB pathway, ROS expression, and decrea-sed endothelial nitric oxide synthesis (93).
Vasculopathy in autoimmune diseases
Autoimmune diseases arise from dysfunction in T cells and B cells that mediate immunosuppression against self-antigens and autoantibody production. As such, the dysfunction in T and B cells prevents the secretion of anti-inflammatory cytokines and promotes a persistent pro-inflammatory state within the body. In systemic lupus erythematosus (SLE), a prototypic multi-organ autoimmune disease, minimal availability of IL-2 leads to decreased survival of regulatory T (Treg) cells (94). The loss of functional Treg cell population induces hyperactivity against otherwise immune-tolerant antigens, causing SLE-associated vasculitis. Moreover, increased level of anti-endothelial cell antibodies in SLE cause endothelial injury by upregulating adhesion molecules and secreting pro-inflammatory cytokines (95). In rheumatic diseases such as systemic sclerosis (SSc) microvascular dysfunction is present in earlier phases of disease. A recent meta-analysis from genome-wide association studies show that epigenetic marks of gene regulation in SSc patients show enrichment for immune imbalance (96). A study by Benyamine et al. (97) showed that SSc patient population presents a subtype of natural killer cells that induce EC activation suggesting the role of dysregulation of immune cells and vasculopathy in SSc.
Tumor-associated angiogenesis
The concept of tumor angiogenesis was first proposed in the 1970s by Folkman (98), who hypothesized that tumors require the formation of new blood vessels to grow beyond a specific size. This theory led to the development of anti-angiogenic therapies for cancer treatment, which aim to disrupt the formation of new blood vessels and limit tumor growth. However, it is now understood that tumor vessels are structurally and functionally abnormal and that tumor-specific ECs play a crucial role in promoting tumor growth and progression through various mechanisms, as described in the previous text. This understanding has led to exploring novel therapeutic strategies targeting tumor-specific ECs for cancer treatment. Tumor-specific ECs express unique markers, such as neuropilin-1 and endosia-lin, absent in normal ECs (99, 100). These markers contribute to the formation of abnormal tumor vessels and promote tumor growth and metastasis. Tumor ECs also express higher levels of adhesion molecules and cytokines, which facilitate the recruitment of immune cells and promote immune evasion by the tumor (101). Additionally, tumor ECs can modulate the tumor microenvironment (TME) by producing growth factors and cytokines that promote tumor cell proliferation and survival.
Tumor angiogenesis plays a crucial role in promoting tumor growth by supplying the tumor with oxygen and nutrients necessary for survival and proliferation. As the tumor grows, it induces the formation of new blood vessels, which are structurally and functionally abnormal and characterized by tumor-specific ECs expressing unique markers and higher levels of growth factors and cytokines (102). Tumor-specific ECs secrete growth factors like VEGF, PDGF, and fibroblast growth factor-2 (FGF-2) to activate downstream signaling pathways involved in EC proliferation and migration, thus promoting angiogenesis in the TME (103-105). In addition, tumor-specific ECs contribute to tumor growth and progression by producing growth factors and cytokines that promote tumor cell survival and prolifera-tion. Thus, the interplay between tumor angiogenesis and tumor growth highlights the importance of targeting tumor-specific ECs for effective cancer therapy. Tumor-specific ECs secrete growth factors, such as VEGF, PDGF, and FGF-2, which promote tumor cell survival and prolifera-tion by binding to their corresponding receptors on tumor cells, such as VEGFR, PDGFR, and FGFR (103-105). Acti-vation of these receptors leads to downstream signaling pathways, including the PI3K-Akt-mTOR and Raf-MEK-ERK pathways, which promote tumor cell survival, proliferation, and angiogenesis (106). Additionally, tumor-specific ECs secrete matrix metalloproteinases that degrade the ECM and facilitate tumor cell migration, further promoting tumor invasion and metastasis (107).
The hypoxic and acidic TME plays a critical role in promoting tumor growth and progression by inducing the expression of genes associated with angiogenesis and tumor growth in tumor-specific ECs. In addition, the acidic environment can activate acid-sensing ion channels on ECs, leading to increased intracellular calcium levels and production of ROS. These changes promote EC proliferation, migration, and angiogenesis, further contributing to tumor growth and progression. Hypoxia-inducible factor 1-alpha, a vital regulator of the cellular response to hypoxia, is also upregulated in tumor ECs under hypoxic conditions, promoting the expression of genes encoding pro-angiogenic factors, such as VEGF and PDGF, and facilitating EC migration and proliferation (108). Targeting these pathways involved in hypoxia and ROS-mediated tumor angiogenesis could represent promising therapeutic strategies for cancer treatment.
Tumor immunotherapy: cancer endothelium as targets
In the last decade, research on cancer immunotherapy has brought about a pivotal change in how we approach the treatment of cancer patients. Novel immunotherapeutic strategies, such as targeting immune checkpoint blockade PD-1/PD-L1, monoclonal antibodies against tumor-associated antigens, and chimeric antigen receptor T cells, have resulted in increased survival rates for patients with otherwise fatal cancers (109, 110). However, cancer immunothe-rapy faces several limitations (111, 112). One notable challenge is that immunotherapy drugs often demonstrate efficacy in only a small subset of cancers, with a response rate of around 15%∼20% in most cases (113). Furthermore, despite the initial strong response to immunotherapeutic drugs, many patients eventually experience a relapse that does not respond to subsequent treatment with immuno-therapeutic drugs.
The endothelium in tumor environments often acts as a selective barrier for immune cell trafficking, hindering the sufficient infiltration of T cells required for effective cancer immunotherapy (114, 115). Angiogenic factors, such as VEGF-A and bFGF, secreted in the TME, lead to reduced expression of VCAM-1 and ICAM-1 on cancer endothelium (116). Additionally, alterations in the chemokine signature within the TME result in decreased trafficking of effector T cells. Nitrosylation of CCL2 through the interaction of ROS prevents CD8+ T cells from interacting with endothelium (117). ECs in the TME also directly express immune inhibitory molecules. Hypoxia and angiogenic factors in the TME increase EC expression of FAS ligands, known to induce T cell apoptosis, and upregulate the expression of PD-L1/2, allowing T cells to evade the TME (118, 119). Recent studies have demonstrated that antiangiogenic therapies enhance T cell adhesion and infiltration into the TME, providing a strong rationale for combinatorial therapeutic strategies (120, 121). These studies have shown that antiangiogenic immunotherapies result in the formation of T cell-enriched tertiary lymphoid-like structures, which are associated with a good prognosis in patients. These structures are surrounded by high endothelial venules (HEVs) that facilitate the infiltration of T cells (121). scRNA-seq of HEVs found in these regions has revealed that antiangiogenic therapies promote the transdifferentiation of postcapillary venules into HEVs (75). Therefore, targeting the cancer endothelium to enhance increased T cell infiltration may be necessary for maximizing the therapeutic effect of current immunotherapies.
Further understanding of EC dysfunction and immune-vascular crosstalk in chronic inflammatory diseases holds promise for advancing the development of more precise and multi-target anti-cancer therapies. ECs play a crucial role in regulating immune responses and vascular homeostasis, and their dysfunction can contribute to the pathogenesis of chronic inflammatory diseases and cancer.
 XML Download
XML Download