

 PDF
PDF Citation
Citation Print
Print



Introduction
The intestinal epithelium is one of the tissues with the most frequent cell turnover (1). Since the identification of intestinal stem cells (ISCs) within its digestive tissue, the fruit fly Drosophila melanogaster has become a valuable model for investigating the regulation of ISCs activity and epithelial tissue turnover (2-4). Leveraging its powerful genetics and experimental tools, the regulation of ISCs activity has been extensively studied (5).
The digestive organ and ISCs in Drosophila share important characteristics with those in mammals (6). ISCs were first identified in the mouse small intestine almost fifty years ago (7). In the mammalian small intestine, spanning the subregions of the duodenum, jejunum, and ileum, the luminal surface is covered with a single layer of epithelium, forming a distinctive crypt-villus architecture (8). ISCs, specifically expressing leucine-rich repeat-containing G-protein-coupled receptor 5 (Lgr5), reside in the bottom of crypts (9). The division of ISCs produces two lineages of progenitors called transit amplifying (TA) cells. These TA cells are committed to differentiating into absorptive enterocytes and other secretory cells, including enteroendocrine cells, respectively (10). The counterpart of the mammalian small intestine in Drosophila is the midgut. Traditionally, the midgut has been subdivided into six regions, R0∼R5, based on anatomical and functional features. Further characterization of the midgut identified as many as 14 subregions based on morphology, histology, and gene expression patterns (5, 11, 12). Like the mammalian small intestine, the Drosophila midgut consists of a single layer of epithelium. However, unlike the crypt-villus structure in mammals, the midgut epithelium is flat. Therefore, Drosophila ISCs are dispersed throughout the epithelium with fewer obvious localization rules.
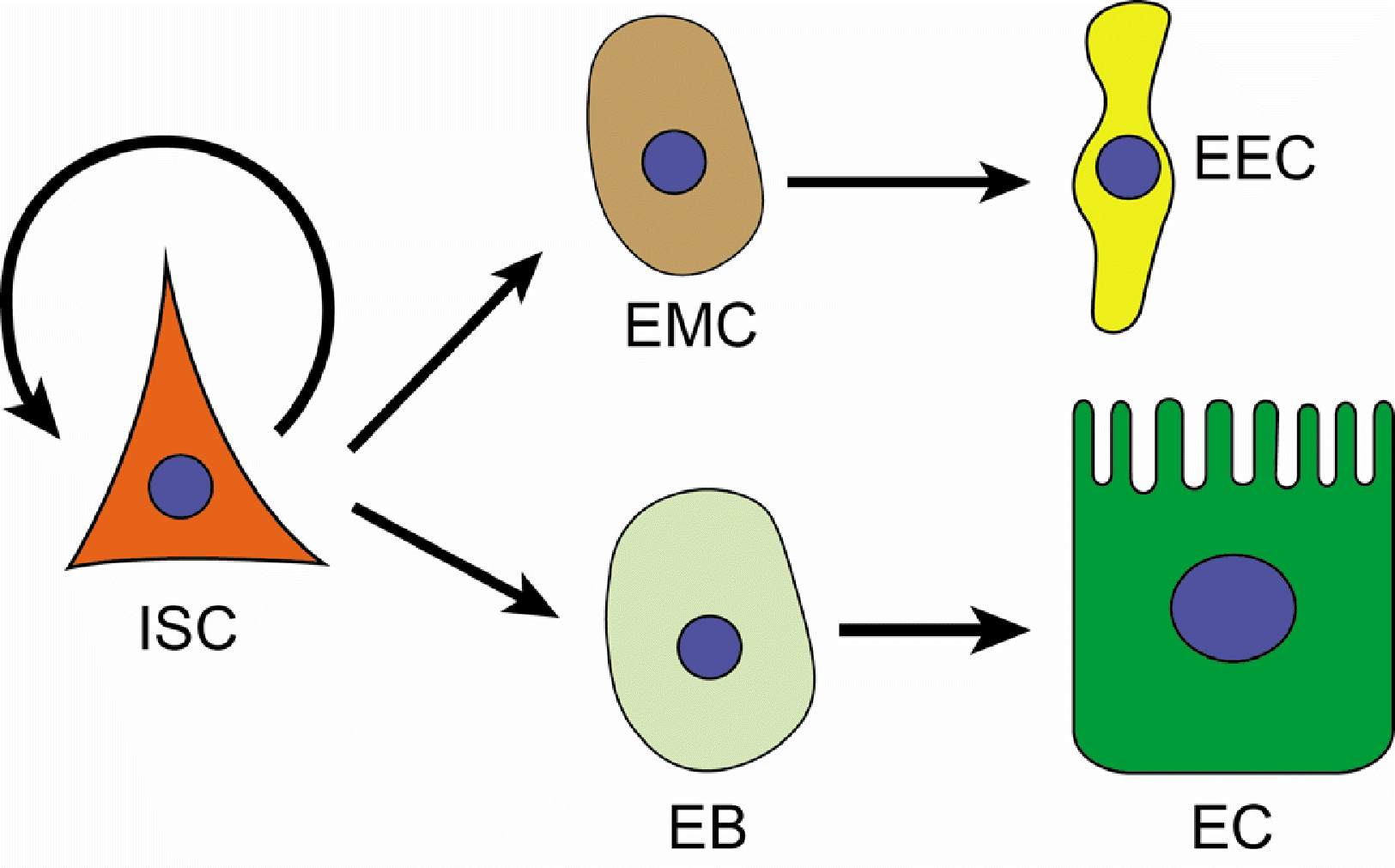
In Drosophila, ISCs divide to give rise to either another ISCs for self-renewal, an enteroblast to be further differentiated into an enterocyte, or an enteroendocrine mother cell capable of producing enteroendocrine cells through cell division (Fig. 1) (13). Comprising approximately 90% of the cells in the epithelium, the enterocyte is the most abundant cell type. These enteric cells constitute a single layer of epithelial tissue. It takes approximately a week to replace the entire tissue with newly proliferated and differentiated enteric cells. However, the rate of ISCs proliferation varies depending on environmental conditions, such as the presence of microbiota and pathogens. Thus, understanding the interaction between enteric cells in the epithelium and such environmental stimuli is crucial for comprehending the physiology and pathology associated with the regulation of ISCs proliferation and differentiation for tissue turnover.
From the environment, Drosophila is constantly in contact with various microbes from indigenous microbiota to harmful pathogens. Drosophila begins to establish its microbiota during the initial interactions with microbes found on eggshells, which are deposited by maternal commensals. Subsequently, the quantity and quality of the microbiota are regulated by various factors, such as microorganisms associated with living environments, feeding behavior, and the host immune systems (14, 15). Typically, the Drosophila gut microbiota consists of 5∼20 species of symbionts primarily belonging to the families Acetobacteraceae and Lactobacillaceae, although the compositions might vary depending on the rearing conditions (14-18). Commensal microbes, especially pathobionts in aging Drosophila are potential activator of tissue turnover process (15). In addition, enteric infections caused by foodborne pathogens represent a significant source of tissue damage, often necessitating the immediate activation of the gut renewal program for survival.
This manuscript is dedicated to discussing the regulation of epithelial tissue turnover in the Drosophila midgut by gut microbes. Here, I have categorized the microbes in contact with the midgut epithelium into two groups: indigenous microbiota and exogenous pathogens, discussing seminal research with a focus on the regulation of tissue turnover.
Microbiota Stimulates Basal Epithelium Turnover
The gut microbiota serves as a crucial regulator of host health. Well-managed microbiota confers various benefits to the host, while dysbiosis has been associated with unfavorable outcomes (19). Emerging research has revealed novel functions of the gut microbiota in influencing diverse aspects of host physiology, spanning from metabolism to behavior (18, 20, 21). These intriguing roles of the microbiota are frequently attributed to specific molecules originating from individual bacterial species (22).
Microbiota for growth promotion through the insulin signal
One of the beneficial roles of gut microbes is to stimulate host growth. While conventionally reared flies typically develop normally even under malnutrition conditions, axenic flies cease to progress beyond the first instar larvae stage and ultimately perish (21). However, this lethality can be rescued through the mono-association of commensal bacteria, such as Acetobacter pomorum. The growth-promoting activity exhibited by A. pomorum appears, at least in part, to be linked to an enhancement of systemic insulin/ IGF-like peptide (DILP) pathway activity. In a separate investigation, the growth-promoting effects of a particular species of commensal bacteria, Lactobacillus plantarum, have also been identified. This species of bacteria was found to improve systemic DILP signal activity, further reinforcing the notion of the role of gut microbes in promoting systemic host insulin signal activity (18). Such studies have highlighted the role of gut microbes in the overall augmentation of host DILP signal activity. However, the precise underlying mechanisms driving this phenomenon remain to be fully elucidated.
Notably, the significance of DILP signal activity in promoting proliferation of ISCs has been emphasized in the context of midgut growth (4). In instances where nutrients are readily available, the local induction of DILP3 occurs from the visceral muscle cells, serving as the niche for ISCs. Subsequently, the activation of the DILP3-induced signaling pathway within the ISCs directs them to divide. Significantly, it is the activity of the DILP3 signal, rather than the availability of nutrients, that primarily governs the regulation of ISCs proliferation (4). The activation of the DILP signal has also been observed in the ISCs undergoing compensatory divisions for homeostasis under stressful conditions (23). This observation is distinct from the ISCs proliferation associated with organ growth, both in terms of the initial stimuli and the subsequent signaling cascades. These findings suggest that signals promoting growth and those triggered by tissue damage converge on a common ISCs proliferation system governed by the DILP signaling pathway. Considering that two major commensal microbes, Acetobacter, and Lactobacillus, reportedly induce DILP signaling, it is plausible that commensal-induced DILP signaling could also influence rates of epithelial cell renewal. Additionally, studies have shown that conventionally reared flies exhibit greater numbers of proliferating ISCs compared to germ-free flies, further emphasizing the influence of gut microbiota on ISCs dynamics (24).
Microbiota, a key player in the IMD pathway-dependent gut homeostasis
The significant influence of the microbiota on the proliferation of ISCs becomes more apparent in pathological conditions characterized by the absence of the IMD pathway, the Drosophila homolog of the mammalian p105-like NF-κB signaling pathway. While conventionally reared wild-type flies demonstrate a modest increase in the rate of ISCs proliferation compared to axenic flies, the rate of ISCs proliferation escalates significantly in flies with mutations in essential components of the IMD pathway, such as Relish or peptidoglycan-recognition protein (PGRP)-LC, leading to an augmented burden of gut microbiota (24). These findings reveal the crucial role of IMD pathway activity in the regulation of microbiota and, consequently, in the process of epithelial renewal.
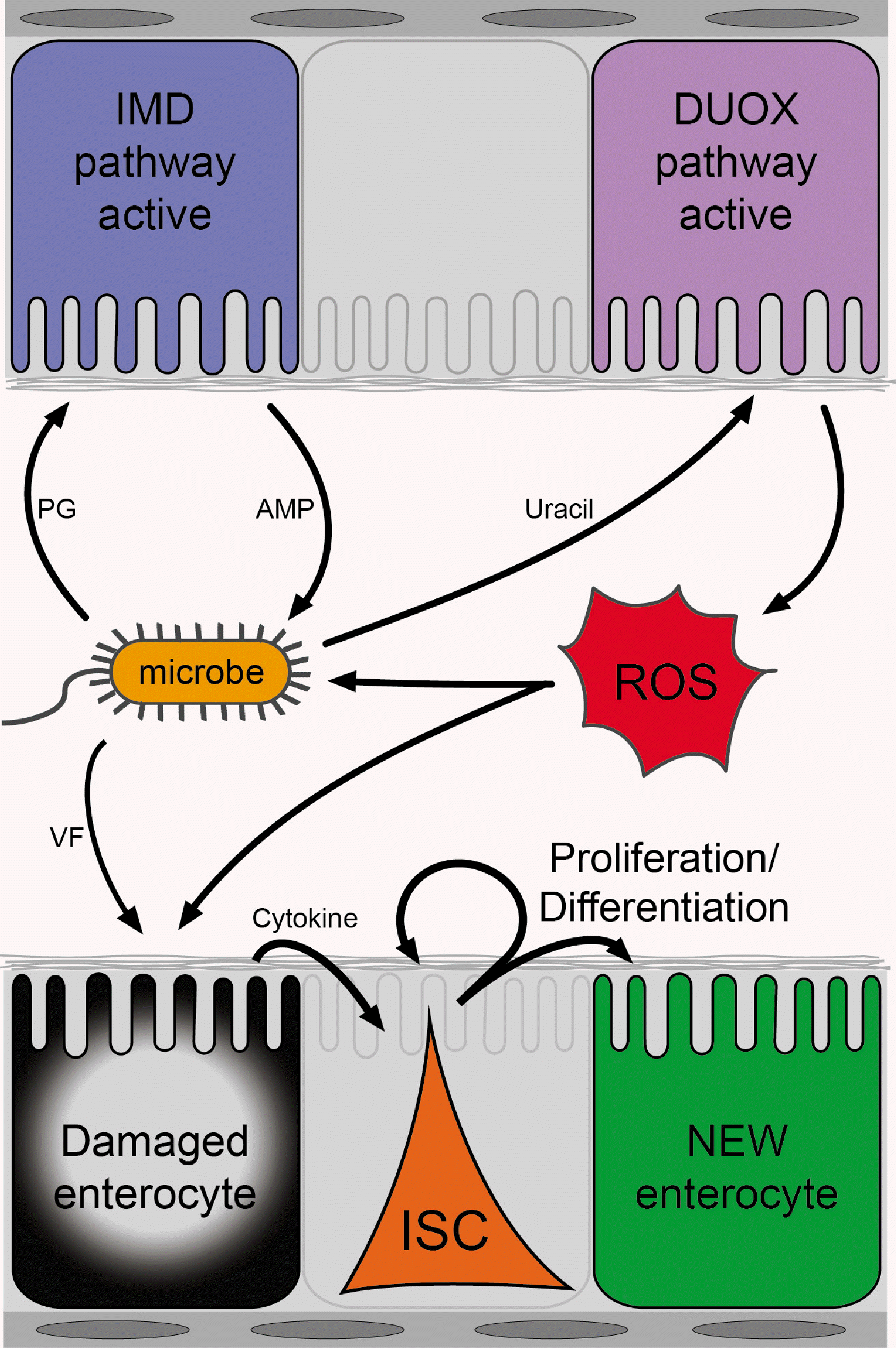
The activation of the IMD pathway represents a crucial innate immune response, initially recognized for its essential role in systemic immunity (25). The pathway is initiated by the recognition of bacterial-derived DAP (meso-diaminopimetic acid)-type peptidoglycans, which constitute the cell wall of gram-negative bacteria. These peptidoglycans are recognized by PGRPs, notably PGRP-LC on the cell membrane and PGRP-LE in the cytoplasm (26-29). Subsequently, the pathway engages intracellular signaling molecules, including IMD and Relish, ultimately culminating in the upregulation of antimicrobial peptides that serve as bactericidal effectors (Fig. 2) (30).
The Drosophila body cavity is typically aseptic, and the induction of the IMD pathway occurs primarily in response to a septic injury. However, within the midgut, the activity of the IMD pathway is predominantly tailored to tolerate commensal microbes, although the pathway can still be induced under conditions such as an enteric infection. To facilitate this regulatory process, the fly gut expresses several inhibitory regulators that serve to curtail the chronic activation of the IMD pathway (5, 15, 31, 32). Consequently, the IMD pathway within the gut is only activated when the stimulus surpasses a certain threshold. Therefore, in the context of the gut epithelium, the activity of the IMD pathway, which does not differentiate between commensals and pathogens (both of which possess peptidoglycans), is likely geared towards controlling the overall load of gut microbes, rather than specifically targeting pathogens. This assertion is supported by the notable increase in the quantity of gut microbes observed when IMD activity is absent (24). Conversely, chronic activation of the IMD pathway leads to alterations in the composition and/or overall load of the microbiota (15, 32). Consequently, the IMD pathway plays a critical role in regulating gut microbes, thereby exerting a significant influence on the ISCs proliferation and the gut repair system.
The role of microbiota in the reactive oxygen species-dependent tissue damage and repair
An increase in the proliferation of ISCs with modified IMD pathway activity is often accompanied by wound responses (24, 32, 33). The activation of various signaling pathways, such as the JNK pathway, in the tissue in response to damage leads to the induction of the expression of cytokines, notably Upd3, which subsequently activates the JAK/STAT signaling in the progenitors (24, 33, 34). The level of Upd3 expression varies depending on the degree of enterocyte stress, thereby fine-tuning the signal activity, and the escalation in the number of dividing stem cells correlates with the augmented expression of upd3 (24). Consequently, the wound responses observed in the context of modified IMD pathway activity and the resulting alterations in the quantity or quality of the microbiota suggest that the microbiota can serve as a source of tissue damage, necessitating compensatory ISCs proliferation (Fig. 2).
How does the microbiota bring about tissue damage and activate the repair system? One possible explanation is that if the microbiota includes symbionts that are more adept at triggering tissue damage than others, an increase in the abundance of these particular symbionts (resulting from either an elevated gut microbe load or a change in their relative proportions) could lead to escalated tissue damage and, consequently, a subsequent elevation in ISCs proliferation. Supporting this model, a study has discovered that L. plantarum is capable of inducing ISCs proliferation (35). L. plantarum produces lactate, which is then oxidized by lactate dehydrogenase to generate NADH. This production of NADH subsequently serves as a fuel source for the generation of reactive oxygen species (ROS) through NADPH-oxidase (NOX). The impact of L. plantarum on ISCs proliferation is minimal in normal flies. However, in mutant animals for PGRP-SD, an upstream signaling component of the IMD pathway, an overgrowth of L. plantarum is observed. In these PGRP-SD flies with impaired IMD pathways, the high numbers of L. plantarum stimulate an excessive production of NOX-dependent ROS, which, in turn, increases the rates of ISCs proliferation, ultimately resulting in hyperplasia (35).
Another study provides additional support for the association between a pathogenic symbiont (referred to as a pathobiont) and tissue damage, particularly in specific animal genotypes such as Caudal mutant animals. Caudal typically functions as a gut-specific transcription repressor for IMD-dependent antimicrobial peptide genes. In animals with a Caudal knockdown, resulting in elevated levels of spontaneous antimicrobial peptide production, there is a significant increase in the prevalence of a pathobiont, Gluconobacter morbifer (15), for reasons currently not fully understood. It has been demonstrated that G. morbifer stimulates dual oxidase (DUOX)-dependent ROS generation, which is subsequently required for the ensuing ISCs proliferation (36). The aforementioned studies shed light on the mechanisms through which indigenous bacteria, particularly under dysbiotic conditions, could contribute to the promotion of damage-associated epithelial turnover.
The DUOX-mediated oxidative burst serves as the primary defense mechanism for gut immunity in Drosophila and simultaneously represents a source of tissue damage (24, 36, 37). The role of DUOX in gut immunity is indispensable, as a reduction in DUOX activity results in lethality following oral infection with Erwinia carotovora supsp. carotovora 15 (Ecc15) or Saccharomyces cerevisiae, which are typically non-lethal in wild-type adult flies (38). However, the DUOX-mediated oxidative burst also serves as a significant source of tissue damage, which can be restored through the process of gut renewal (24). These findings underscore the critical importance of tightly regulating this pathway.
An intriguing aspect of the DUOX pathway pertains to its interaction with a bacterial-derived metabolite, uracil. Ecc15 triggers DUOX activation in the gut lumen by secreting uracil (22). The secretion of uracil and the subsequent activation of the DUOX pathway have also been observed in the pathobiont G. morbifer, the entomopathogen Pseudomonas entomophila, and various human pathogens, including Pseudomonas aeruginosa, Serratia marcescens, and Enterococcus faecalis. In contrast, other symbionts are incapable of producing uracil and therefore maintain DUOX in an inactive state (Fig. 2) (22). Recent research has identified that uracil is a product of bacterial nucleoside catabolism by pathogens and pathobionts that utilize uridine (39). These microbes convert uridine into ribose and uracil. Subsequently, ribose is utilized for quorum sensing and virulence gene expression, while uracil is excreted. These findings support the hypothesis that the metabolic activities of gut microbes represent crucial components in the intricate interplay between the host and microbiota, thereby influencing tissue homeostasis. This concept is further reinforced by the discovery demonstrating that lactate derived from L. plantarum also stimulates the generation of ROS in a dysbiotic condition via the intestinal NOX (35).
Interestingly, the activity of DUOX is inherently influenced by the metabolic status of the host tissue itself, even in the absence of infection (40). Specifically, genetic manipulations leading to enterocyte lipolysis activate the DUOX pathway, whereas increased lipogenesis results in DUOX inactivation. The impact of lipid metabolism on the regulation of DUOX activity is attributed to the observation that lipolysis produces NADPH, which serves as a substrate for DUOX. These findings propose a fascinating hypothesis suggesting that infected cells reprogram their metabolic status towards lipid catabolism to augment the immunological output, particularly the oxidative burst.
ROS, a mediator for ISCs proliferation
Thus far, our discussion has centered around a model highlighting the tissue repair system induced by damage. In specific scenarios, gut microbes activate the DUOX pathway-dependent host immune system, leading to the excessive production of ROS. Subsequently, these ROS act on the epithelial cells, instigating damage-dependent signals that promote the proliferation of ISCs. However, evidence suggests that ROS can also function as a signaling molecule for stem cell activity by triggering multiple signaling pathways (41). For instance, in the Drosophila lymph gland, a specialized organ responsible to produce blood cells, ROS serves as an intrinsic factor for the differentiation of hematopoietic progenitors (42). Additionally, in the Drosophila gut, the proliferation of ISCs is directly proportional to the levels of ROS, as demonstrated when the ROS levels were manipulated either by downregulating the mitochondrial electron transport chain or by overexpressing antioxidant genes in the progenitors (43). Studies have shown that CncC, a transcription factor that induces the expression of antioxidant genes, acts in opposition to the proliferation of ISCs, further supporting the notion that ROS serves as a regulator for ISCs proliferation (43). A recent investigation has also highlighted the critical role of the calcium signal in ISCs in transducing signals from oxidative stress to ISCs proliferation (44). Therefore, as a multifunctional agent, ROS may act on various types of cells, eliciting cellular signals that ultimately culminate in the mitogenic activity of ISCs. Further research is warranted to gain a more nuanced understanding of the diverse functions of ROS.
Pathogens Trigger Homeostatic Tissue Repair
The oral infection of pathogens has served as a valuable model system for investigating tissue renewal mechanisms. Several Drosophila pathogens, such as Ecc15, P. entomophila, and S. marcescens, have been utilized to explore their association with the tissue repair system.
Erwinia carotovora supsp. carotovora 15
Ecc15 is a Gram-negative phytopathogen that is transmitted through insect vectors (45). Ecc15 is equipped with a virulence factor known as the Erwinia virulence factor, which contains a palmitoyl moiety, although its complete function remains yet to be fully understood (46, 47). Upon enteric infection, Ecc15 releases uracil, thereby triggering a local DUOX-dependent immune response, akin to the mechanism observed in the pathobiont G. morbifer (22, 36). The activation of DUOX leads to tissue damage, which is subsequently countered by compensatory ISCs proliferation (24). The observed increase in the number of mitotic ISCs following Ecc15 infection can be attributed to the DUOX-mediated host defense system, as evidenced by the failure of ISCs proliferation to increase even in the presence of Ecc15 infection when DUOX activity is compromised (24). Thus, findings derived from studies using Ecc15 underscore the role of immunopathology stemming from host-generated ROS as a significant contributor to gut damage, thereby necessitating the tissue renewal process (Fig. 2).
Pseudomonas entomophila
P. entomophila is a Gram-negative entomopathogen that can induce lethality in Drosophila when ingested at high doses, thereby disrupting tissue homeostasis (24, 48). P. entomophila relies on specific virulence factors for successful infection, including a pore-forming toxin known as Monalysin (49, 50). The gagA:gagS two-component system is essential for the production of these virulence factors, and a gagA mutant strain of P. entomophila has been found to be avirulent (49). Intriguingly, while the ingestion of a low dose of P. entomophila promotes the proliferation of ISCs, flies ingesting a high dose of P. entomophila do not exhibit any significant signs of gut repair, suggesting that the repair program can be inhibited when the extent of damage exceeds a certain threshold (24). Subsequent investigations revealed that, although flies fed a high dose of P. entomophila do express mRNAs related to damage-dependent signaling genes, global translation is impaired (51). The evidence suggests that an excessive level of oxidative stress is responsible for the blockade of global translation and subsequently the repair system. This hypothesis is supported by several key observations. Firstly, the mitigation of oxidative stress in P. entomophila-infected flies, either through co-feeding with antioxidants or via genetic manipulation to reduce DUOX activity, leads to an enhancement in translation. Additionally, the co-feeding of Ecc15 and paraquat, a potent ROS inducer, resulted in a decrease in translation compared to that observed with Ecc15 alone (51). These findings underscore the significance of immunopathology following local pathogen infection and emphasize the indispensable role of the tissue repair system for the survival of the host organism.
Serratia marcescens
S. marcescens is a human opportunistic pathogen known to infect Drosophila, displaying several intriguing characteristics (52-54). Upon ingestion, S. marcescens secretes various virulence factors, including hemolysin with pore-forming activity (55). An early response from the host following infection is the extrusion of cytoplasm from enterocytes into the lumen, which serves as a mechanism to mitigate toxicity (56). Furthermore, S. marcescens triggers damage-dependent host signaling pathways, such as the JAK/STAT signal activity in the gut epithelium. The activation of these damage-dependent signals in response to S. marcescens infection ultimately promotes compensatory ISCs proliferation (57-59).
Concluding Remarks
In this manuscript, microbiota and pathogens interacting with the intestinal epithelium of Drosophila have been discussed in the context of ISCs regulation and tissue turnover. Each microbe employs a distinct strategy to interact with the host. Pathogens utilize the virulence factors unique to each bacterial species contribute to varying pathological outcomes. However, investigations utilizing Drosophila pathogen models have consistently revealed a common theme: microbial infections trigger the activation of the host’s immune system, leading to the utilization of bactericidal ROS. While ROS serves as the primary source of tissue damage, it also functions as a potential signaling molecule that triggers the division of ISCs. Consequently, the resistance mechanism engaged to eliminate invading pathogens comes at the cost of tolerating tissue damage, thereby necessitating the activity of the tissue repair system for the survival of the host organism. Enhanced resistance may strengthen the immune function but exacerbate tissue damage, whereas heightened tolerance may result in hyperplasia or cancer due to excessive stem cell activity, representing an additional detrimental outcome (35, 57).
 XML Download
XML Download