

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Normal T cells can recognize tumor-associated antigens (TAAs) and produce an antitumor immune response as a normal body defense, depending on antigen presentation by major histocompatibility complexes (MHCs). However, tumor cells can downregulate their expression of MHCs and escape the antitumor effect of the T-cell-mediated immune reaction. To overcome this limitation of normal T cells, autologous T cells are genetically engineered into chimeric antigen receptor (CAR) T cells that have an extracellular antigen-recognition domain, such as B cells, and an intracellular signaling domain, such as activated T cells [1, 2]. The antigen-recognition domain most commonly consists of a single-chain variable fragment domain that binds to TAAs. Thus, according to the target TAAs, diverse CAR T cells can be produced against various malignancies, including solid and blood cancers. Preclinical and clinical investigations of the efficacy of various CAR T cells for solid cancers are ongoing. The currently approved CAR T cells target two antigens in B-cell lymphoid malignancies: CD19 and B-cell maturation antigen (BCMA). The intracellular signaling domains are necessary for the activation of T cells and consist of a costimulatory domain from proteins such as CD28 or 4-1BB (CD137) and an intracellular CD3ζ activation domain. The currently used second-generation CAR T cells have one costimulatory domain from CD28 or 4-1BB, whereas third-generation CAR T cells have two costimulatory domains [3]. Through this genetic modification, CAR T cells with an extracellular domain targeting a specific TAA can generate an antitumor effect in an MHC-independent manner via the interaction between their extracellular domain and a target TAA. In this review, we summarize the issues with currently approved CAR T cells for patients with relapsed or refractory B-cell lymphoid malignancies, focusing on toxicities after CAR T-cell therapy and post-CAR T-cell failure.
Go to : 
Current CAR T-cell therapies
The Food and Drug Administration (FDA) has approved four anti-CD19 CAR T-cell products, tisagenlecleucel (tisa-cel), axicabtagene ciloleucel (axi-cel), lisocabtagene maraleucel (liso-cel), and brexucabtagene autoleucel (brexu-cel) for patients with relapsed or refractory (r/r) B-cell lymphoma (BCL), and two anti-BCMA CAR T-cell products, idecabtagene vicleucel (ide-cel) and ciltacabtagene autoleucel (cilta-cel), for the treatment of r/r multiple myeloma (MM) (Fig. 1). Tisa-cel, the first FDA-approved CAR T-cell product, uses 4-1BB as a costimulatory domain. The indications for tisa-cel are as follows: 1) patients >18 yrs with r/r diffuse large B-cell lymphoma (DLBCL) after two lines of therapy, based on the JULIET trial reporting a 52% overall response rate (ORR) and a 40% complete response (CR) rate [4]; 2) patients >25 yrs with r/r B-acute lymphoblastic leukemia (ALL), based on the ELIANA trial reporting an 81% ORR [5]; and 3) patients >18 yrs with r/r follicular lymphoma (FL) grades I–IIIA after two lines of therapy or who relapsed after autologous stem cell transplantation, based on the ELARA trial reporting a 69% CR rate and a 86% ORR [6].
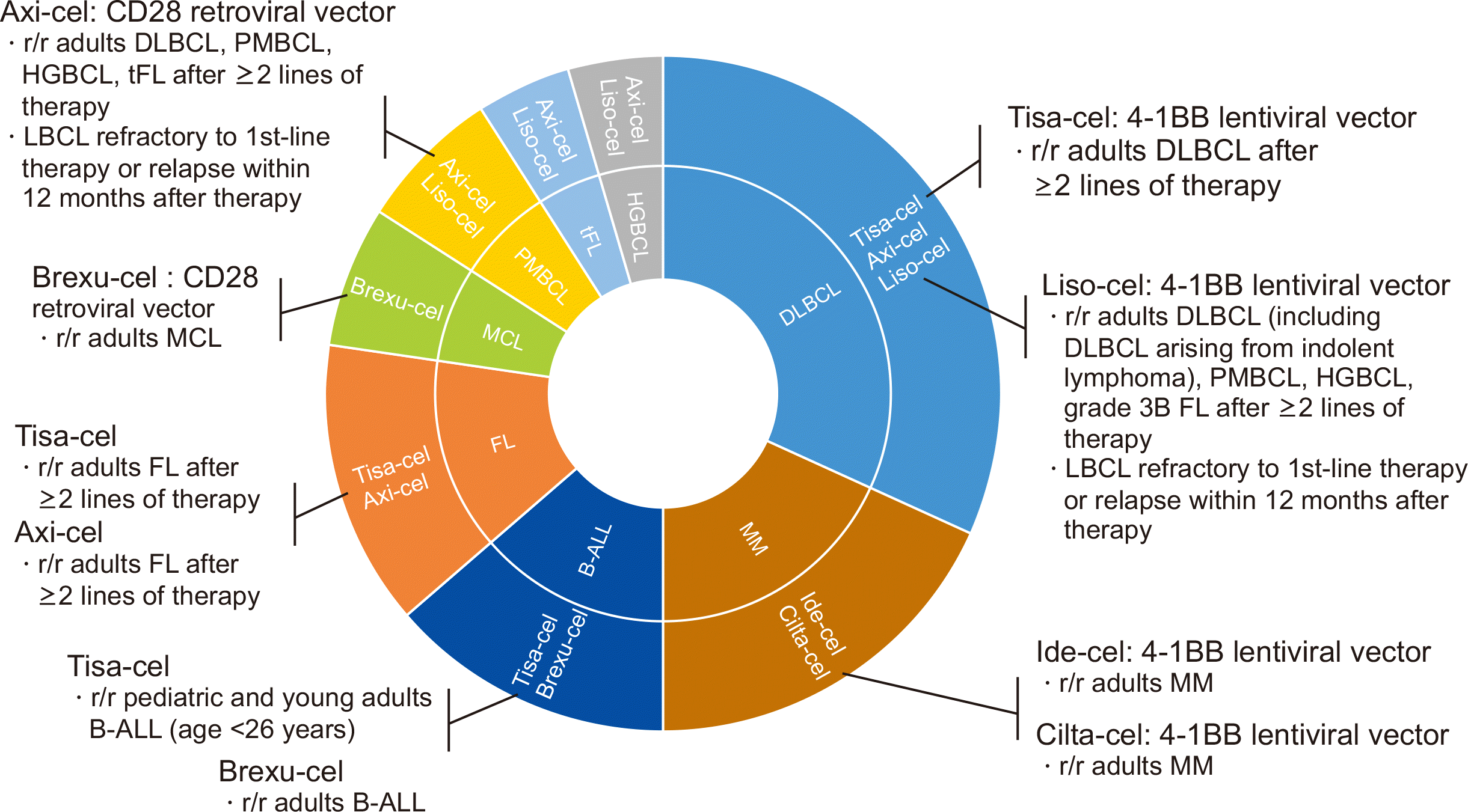 | Fig. 1Currently approved CAR T-cell products and their indications.
Abbreviations: CAR, chimeric antigen receptor; r/r, relapsed or refractory; DLBCL, diffuse large B-cell lymphoma; HGBCL, high-grade B-cell lymphoma; PMBCL, primary mediastinal large B-cell lymphoma; MCL, mantle cell lymphoma; FL, follicular lymphoma; tFL, transformed follicular lymphoma; B-ALL, B-acute lymphoblastic leukemia; MM, multiple myeloma.
|
The second FDA-approved CAR T-cell product, axi-cel, uses the costimulatory domain of CD28 and has the following indications: 1) patients >18 yrs with r/r large BCL, including DLBCL, after at least two lines of therapy, based on the ZUMA-1 trial reporting an 82% ORR and 54% CR rate [7]; and 2) patients >18 yrs with r/r FL grades I–IIIA after at least two lines of therapy, based on the ZUMA-5 trial reporting a 94% ORR and 79% CR rate [8]. Brexu-cel has the same structure as axi-cel; however, circulating CD19-positive tumor cells are removed to prevent the premature activation of CAR T cells during manufacturing.
Brexu-cel has the following indications: 1) patients >18 yrs with r/r B-ALL, based on the ZUMA-3 trial reporting a 71% CR rate [9]; and 2) patients with r/r mantle cell lymphoma, based on the ZUMA-2 trial reporting a 93% ORR and a 67% CR rate [10]. Liso-cel is another 4-1BB-based CAR T-cell with a 1:1 ratio of CD4 and CD8 T cells and is indicated for patients >18 yrs with r/r large BCL, including DLBCL, based on the TRANSCEND NHL 001 trial reporting a 73% ORR and a 53% CR rate [11].
Axi-cel and liso-cel are also prescribed to patients with large BCL who are either refractory to first-line immunochemotherapy or relapse within 12 months of first-line immunochemotherapy because of their superior outcomes compared with those seen in patient who underwent conventional salvage chemotherapy in the ZUMA-7 and TRANSFORM trials [12, 13]. For the treatment of r/r MM, two BCMA-directed CAR T cells with a costimulatory domain, 4-1BB, have been approved. Ide-cel, the first BCMA-directed CAR T-cell product, was approved based on the KarMMA trial reporting a 73% ORR in patients receiving at least three previous regimens, including a proteasome inhibitor, an immunomodulatory agent, and an anti-CD38 antibody [14]. Cilta-cel was approved for the same indication, based on the CARTITUDE-1 trial reporting a 97% ORR and a 77% 12-month progression-free survival (PFS) [15].
Go to :
Challenging issues in CAR T-cell therapy
The manufacturing process of CAR T cells comprises multiple steps. In the first step, T cells are collected from patients via apheresis (Fig. 2). The collected T cells are transferred to the manufacturing facility, where they are virally transduced with CAR, followed by the ex vivo expansion of the CAR-transduced T cells. Manufacturing CAR T cells generally takes 2–4 weeks. The CAR T cells are transferred to the hospital, where they are infused into the patient. This process has a long vein-to-vein time, extending to over six weeks, because of the turnaround and transportation times. Accordingly, patients with unstable disease may not be appropriate for current CAR T-cell therapy because they may not endure the long vein-to-vein time given their deteriorating disease status.
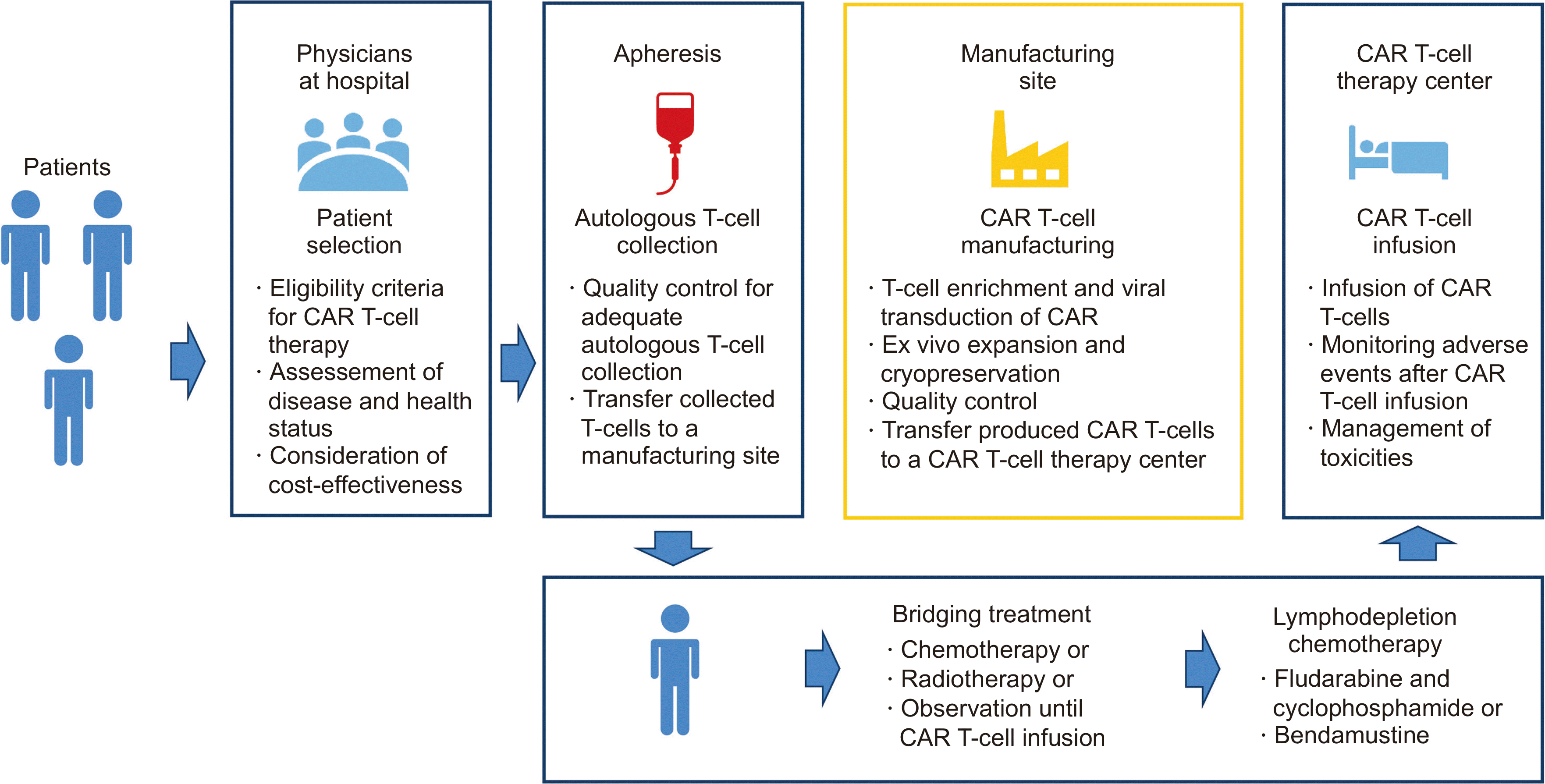 | Fig. 2Steps in the process of CAR T-cell therapy. (1) Physicians should select patients who are appropriate for CAR T-cell therapy. (2) Autologous T cells should be collected via a qualified process. (3) Patients may need various bridging therapies during CAR T-cell manufacturing. (4) Patients receive CAR T cells after lymphodepletion chemotherapy, and the CAR T-cell therapy center should establish standardized protocols for monitoring and managing toxicities after CAR T-cell therapy.
Abbreviation: CAR, chimeric antigen receptor.
|
A recent multicenter retrospective observational study reported inferior survival outcomes in European patients compared with those in US patients because of the longer time-to-infusion interval in Europe [16]. In the second step, CAR T cells, such as tisa-cel and liso-cel, can be produced from frozen autologous T cells. Thus, patients living in a country with no manufacturing facility can receive CAR T-cell therapy. However, axi-cel and brexu-cel can only be produced in countries where manufacturing facilities are established because they are manufactured from fresh autologous T cells. Therefore, logistic hurdles that prevent timely access to CAR T cells remain, and there are limitations in selecting CAR T cells according to the region where the patient lives [17].
In the third step, bridging chemotherapy is administered to most patients while waiting during the turnaround time for CAR T-cell manufacturing and delivery. A recent real-world study by the German Registry for Stem Cell Transplantation reported that the 12-month PFS of patients responding to bridging therapy was significantly better than that of patients with no response to bridging therapy (53% vs. 20%, P<0.001) [18]. Although the optimal regimen for bridging therapy in terms of intensity and efficacy is unclear [19, 20], a recent retrospective study of 375 patients in the UK corroborated that a complete or partial response to bridging therapy reduced the risk of progression or death after CD19 CAR T-cell therapy and that bridging therapy with polatuzumab vedotin may improve outcomes of CAR T-cell therapy, particularly tisa-cel [21].
In the fourth step, before CAR T-cell infusion, patients should receive lymphodepletion chemotherapy because priming the immune environment is important for successful CAR T-cell therapy. Lymphodepletion can promote a favorable cytokine profile, including increased interleukin (IL)-7 and IL-15 levels, leading to the expansion and persistence of the infused CAR T cells [22]. Although the optimal regimen for lymphodepletion chemotherapy has not been determined, regimens incorporating fludarabine and cyclophosphamide are commonly used.
In the final step, it is important to select patients who are appropriate for CAR T-cell therapy considering the benefits and risks, as well as the high cost of and limited resources for this treatment [23]. For heavily pretreated patients, there may be a risk of manufacturing failure, and patients with a high tumor burden may not benefit from CAR T-cell therapy.
Go to :
Cytokine-related toxicities after CAR T-cell therapy
Cytokine release syndrome (CRS) is the most common adverse event in patients receiving CAR T-cell therapy, and the onset of CRS following CAR T-cell infusion can vary from hours to 1–3 days [24, 25]. Various factors, including tumor burden, target disease, and pre-CAR T-cell health status, can influence the occurrence of CRS. The onset of CRS is closely related to the type of costimulatory domain of CAR T cells [26]. Axi-cel typically induces CRS earlier than tisa-cel because CD28 causes a more rapid systemic inflammatory reaction than 4-1BB [27]. Inflammatory cytokines released by activated immune cells, including monocytes/macrophages, can cause CRS, and IL-6 produced by circulating monocytes is a key cytokine inducing CRS after CAR T-cell therapy [28]. Tocilizumab, a humanized anti-IL-6 receptor monoclonal antibody of the IgG1 class, was used in the first pediatric patient with r/r B-ALL who received CD19 CAR T cells because she developed CRS [29]. Tocilizumab inhibits IL-6-mediated signal transduction by blocking IL-6 binding to its receptor [30]. This led to its FDA approval for the treatment of CAR T-associated CRS, and the recommended dose is 4–8 mg/kg (maximum, 800 mg) [31]. However, the clinical manifestations and laboratory findings of CRS can overlap with those of other inflammatory conditions related to sepsis or tumor lysis syndrome. Accordingly, other clinical conditions mimicking CRS should be excluded, and CRS must be confirmed before tocilizumab treatment. Currently, there is no established biomarker for predicting severe CRS. Given the negative impact of severe CRS on CAR T-cell therapy outcomes, treatment with tocilizumab may be considered for CRS of grade 1 or higher, and our institution uses tocilizumab as early as possible in clinical practice based on recommended guidelines (Table 1) [32, 33].
Table 1
Modified protocol for assessing and managing cytokine-related toxicities after CAR T-cell therapy at the Samsung Medical Center based on the ASTCT and ASCO guidelines
Abbreviations: ASCO, American Society of Clinical Oncology; ASTCT, American Society for Transplantation and Cellular Therapy; BiPAP, bilevel positive airway pressure; CAR, chimeric antigen receptor; CPAP, continuous positive airway pressure; CRS, cytokine release syndrome; GCSF, granulocyte colony-stimulating factor; ICANS, immune effector cell-associated neurotoxicity syndrome; ICE, immune effector cell-associated encephalopathy; ICU, intensive care unit; IV, intravenous.
![]()
Immune effector cell-associated neurotoxicity syndrome (ICANS) is another unique adverse event after CAR T-cell therapy. ICANS is rarer than CRS, and any newly occurring neurological manifestations after CAR T-cell therapy, including aphasia, headache, encephalopathy, focal neurological deficits, tremors, and seizures without any other cause, can be designated as ICANS. The onset of ICANS varies from within a few hours to more than 2–3 weeks after infusion. Although ICANS can occur without an event of preceding CRS, most cases of ICANS occur following CRS, and its severity is related to the degree of CRS [34, 35].
Although the pathogenesis of ICANS has not been fully identified, increased cytokine levels in the central nervous system (CNS) owing to the breakdown of the blood–brain barrier (BBB) and increased vascular permeability may be the main triggering factors for ICANS [36]. Therefore, various cytokines may be related to ICANS, including interferon (IFN)-γ, granulocyte-macrophage colony-stimulating factor (GM-CSF), IL-1, IL-6, IL-10, and IL-15 [28, 37]. Although the evidence is still weak, the penetration of CAR T cells into the CNS may be associated with the occurrence of neurotoxicity because increased CAR T-cell levels in the cerebrospinal fluid have been reported in ICANS [38]. Although the causative factors for ICANS remain unclear, patients with neurological comorbidities or a high tumor burden before CAR T-cell therapy may be at risk of ICANS [34, 36]. The number of infused CAR T cells and their peak levels after infusion may also be related to ICANS [39, 40], and the type of costimulatory molecule may affect ICANS, as its incidence is lower with 4-1BB than with CD28 [34, 36]. The role of tocilizumab in treating ICANS is limited because it is less effective in ICANS than in CRS. Tocilizumab has poor BBB permeability, and IL-6 levels in the CNS may increase as a compensatory reaction to the administration of tocilizumab [41]. Therefore, when ICANS occurs after or simultaneously with CRS, tocilizumab can be used; however, systemic dexamethasone should be considered immediately after ICANS is identified. As a first-line treatment for ICANS of grade ≥2, dexamethasone 10 mg should be administered every 6–8 hrs [42, 43]. In cases where dexamethasone is ineffective, or ICANS becomes grade 4, the intravenous administration of methylprednisolone 1 g is recommended and then slowly tapered because rapid reduction could aggravate ICANS; however, the optimal dose and duration of the corticosteroid remain uncertain [43]. There are some concerns regarding the negative effect of the corticosteroid on the efficacy of CAR T-cell therapy, although the accumulating data do not show an association between corticosteroid use and poor outcomes of CAR T-cell therapy [40, 44]. As there are currently no established alternative agents for steroid-refractory ICANS, early detection and immediate treatment may be the best approach for managing ICANS (Table 1). Anakinra, an IL-1 receptor antagonist, and siltuximab, an anti-IL-6 antibody, have been suggested for the treatment of ICANS because monocyte-derived IL-1 signaling can affect the occurrence of ICANS, and active IL-6 in CNS may also play a role in its occurrence [28]. In a recent single-center study, anakinra showed efficacy for steroid-refractory ICANS [45]. Siltuximab binds to circulating IL-6, thereby reducing it, and may be a salvage therapy for patients who do not respond to tocilizumab or corticosteroids [46].
Another issue is severe toxicity resembling hemophagocytic lymphohistiocytosis (HLH) after CAR T-cell infusion, so-called CAR-HLH or immune effector cell-associated hemophagocytic lymphohistiocytosis-like syndrome [47-49]. HLH is an uncontrolled hyperinflammatory syndrome characterized by excessive macrophage and lymphocyte activation and pro-inflammatory cytokine production [50, 51]. The clinical features and laboratory findings of HLH resemble those of CRS, including fever, cytopenia, and multiorgan failure. The reported incidence of CAR-HLH is only 1%–3%; however, the prognosis is poor, and the mortality rate is up to 80% [27, 52]. Currently, the diagnosis of secondary HLH is based on the HLH-2004 criteria, including increased ferritin, bone marrow hemophagocytosis, triglycerides, and soluble IL-2R [53]. However, these criteria are not specific, and patients with lymphoid malignancies may show similar clinical and laboratory features regardless of the history of CAR T-cell therapy [54, 55]. Accordingly, diagnosing CAR-HLH requires new diagnostic criteria in the context of CRS. The National Comprehensive Cancer Network guidelines propose to consider HLH after CAR T-cell infusion in case of rapidly increasing and high serum ferritin levels (>5,000 ng/mL) with cytopenia in the context of fever, especially if accompanied by any of the following: 1) Common Terminology Criteria for Adverse Events (CTCAE) grade ≥3 increase in serum bilirubin, AST, or ALT levels; 2) CTCAE grade ≥3 oliguria or increased serum creatinine levels; and 3) CTCAE grade ≥3 pulmonary edema [56]. Histological demonstration of hemophagocytosis in bone marrow or organs based on histopathological assessment of cell morphology and CD68 immunohistochemistry also indicates CAR-HLH. Considering the dismal prognosis of patients with CAR-HLH, more active therapy with tocilizumab and steroids should be promptly started for patients with suspected HLH, as in the management of high-grade CRS (Table 1). In refractory cases, the administration of etoposide and other agents recommended for adult HLH, including the IFN-γ inhibitor emapalumab, can also be considered, although data on this treatment are scarce [57, 58].
Go to :
Hematological toxicities and infections after CAR T-cell therapy
Hematological toxicities, particularly neutropenia, are frequently reported following CAR T-cell infusion across various disease entities and CAR T-cell types [24, 59]. Although most patients experience cytopenia after CAR T-cell infusion, it is usually transient and related to the myelosuppression of lymphodepletion chemotherapy. However, a second episode of cytopenia, the so-called second dip, can occur and last for months and even years after CAR T-cell infusion [60]. Although the underlying mechanism of the second dip has not been identified, the interaction of CAR T cells with tumor cells, resulting in systemic inflammatory stress, including CRS and oligoclonal T-cell expansion, may influence the occurrence of cytopenia [61, 62]. Furthermore, the baseline hematopoietic reserve before CAR T-cell infusion may be associated with prolonged cytopenia. The recently developed risk model, CAR-HEMATOTOX, consists of the hematopoietic reserve (absolute neutrophil count, Hb, and platelet count) and baseline inflammation (C-reactive protein and ferritin levels) [63]. This model relying on laboratory values before lymphodepletion chemotherapy aims to predict the risk of developing severe hematologic toxicity after CAR T-cell therapy (Fig. 3). Notably, high CAR-HEMATOTOX risk is associated with substantial cytopenia, severe infections, and prolonged hospitalization [64].
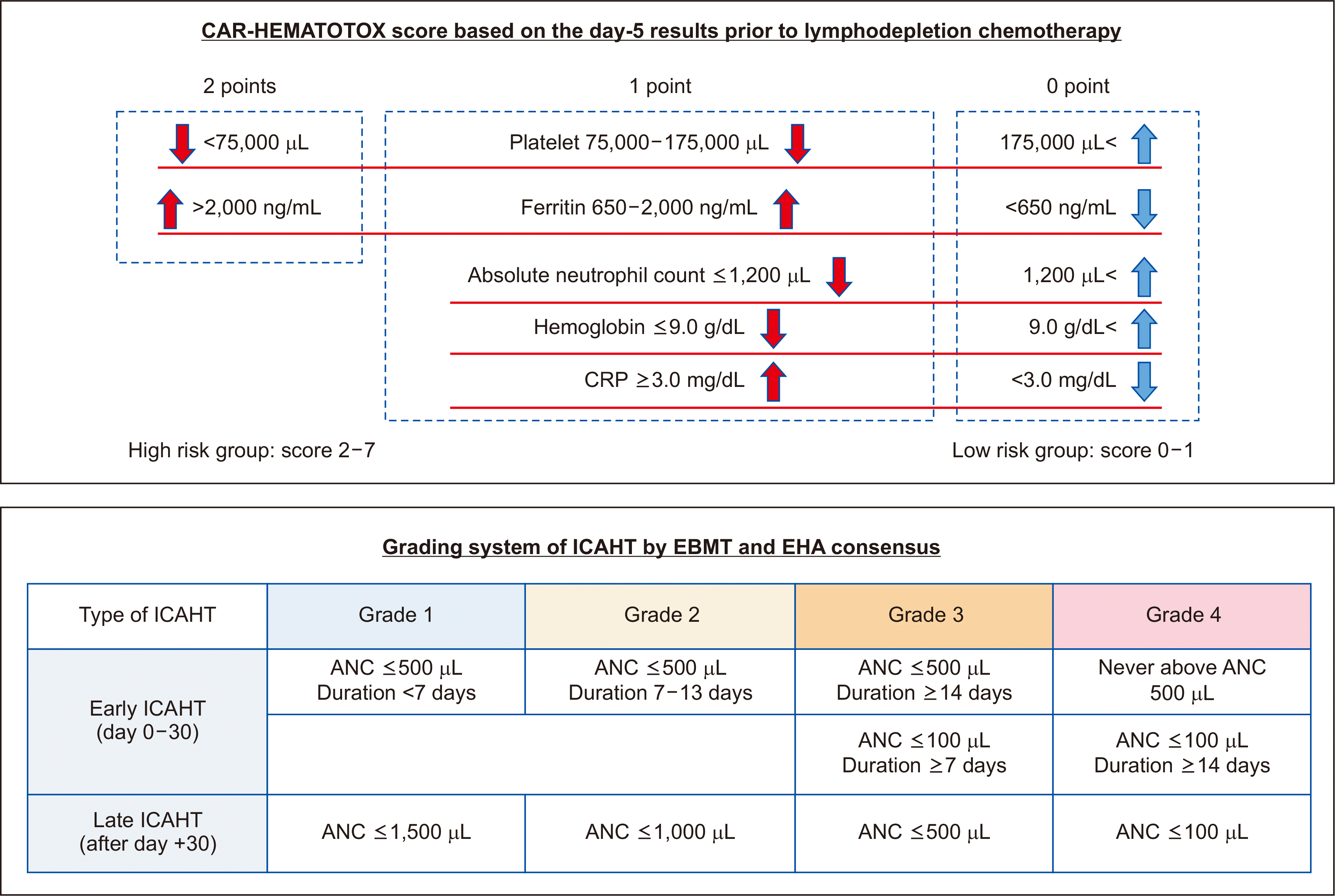 | Fig. 3Guidance for assessing hematological toxicity after CAR T-cell therapy. In the CAR-HEMATOTOX model, low platelet count, high ferritin level, low absolute neutrophil count, low Hb level, and high CRP level are risk factors for hematologic toxicity. Based on the sum of scores, high or low risk can be determined. Based on the ICAHT grading system, early and late ICAHT can be determined according to the time after CAR T-cell infusion.
Abbreviations: CAR, chimeric antigen receptor; ICAHT, immune effector cell-associated hematotoxicity; EHA, European Hematology Association; EBMT, European Society for Blood and Marrow Transplantation; CRP, C-reactive protein; ANC, absolute neutrophil count.
|
As the clinical course of hematological toxicity after CAR T-cell therapy differs from that of cytopenia after conventional chemotherapy, it was named immune effector cell-associated hematotoxicity (ICAHT), and a recent consensus by the European Hematology Association (EHA) and European Society for Blood and Marrow Transplantation (EBMT) recommends a new grading system according to the onset of ICAHT [65]. The expert panel, on behalf of the EHA and EBMT, defined early and late ICAHT as the occurrence of cytopenia during the first 30 days and 30 days after CAR T-cell infusion, respectively. A grading system based on both the depth and duration of neutropenia was defined for early ICAHT, whereas late ICAHT was graded based on the depth of neutropenia (Fig. 3). Given the negative impact of early neutropenia on CAR T-cell therapy outcomes, early granulocyte CSF (GCSF) administration on day +2 can be considered for patients with high CAR-HEMATOTOX risk to shorten the duration of neutropenia. Although GM-CSF should be avoided because of its relation with inflammatory toxicity, early use of GCSF reportedly reduces febrile neutropenia without increasing the risk of CRS or ICANS and a negative impact on CAR T-cell therapy response and outcomes [66, 67]. Based on patient risk and institutional standards, the expert panel recommended the existing grading, such as CTCAE and transfusion, for anemia and thrombocytopenia.
Infection is the leading cause of nonrelapse mortality (NRM) in patients receiving CAR T-cell therapy, and the pattern of infections changes over time, as in stem cell transplantation. The LYSA study from the DESCAR-T registry reported 48 cases (5.0% of all patients) of NRM among 957 patients who received CD19 CAR T cells in 27 French centers, and infections accounted for 56% of all cases (29% non-coronavirus disease 2019 (COVID-19) infections, 27% COVID-19 infections) [68]. Managing COVID-19 and other viral infections is an important and challenging issue in patients with hematologic malignancies, including CAR T-cell recipients [69, 70]. As the prevalence of COVID-19 declines, the risk of COVID-19 may change over time, but it remains a problematic issue in the management of post-CAR T-cell therapy. Considering the risk of infectious complications after CAR T-cell therapy, antiviral and antipneumocystis agents should be implemented for all patients, and bacterial and fungal prophylaxis should also be considered according to the risk for each patient.
Go to :
Progression or relapse after CAR T-cell therapy
Although CAR T-cell therapy has shown better efficacy than conventional salvage therapies, disease progression or relapse after CAR T-cell infusion is a concern. Suggested mechanisms of resistance and relapse after CAR T-cell therapy include several aspects. First, tumor factors can influence treatment failure after CAR T-cell therapy. Target antigen loss after CAR T-cell therapy can be related to resistance to CAR T-cell therapy. For example, a patient with DLBCL showed loss of CD19 on the surface of tumor cells at the time of disease progression two months after CD19 CAR T-cell infusion [71]. Genetic changes in CD19, such as deletions, frameshifts, and other mutations, may result in the loss of the CD19 antigen in tumor cells. However, the preexistence of CD19-negative tumor cells can also account for CD19-negative relapse because preexisting CD19-negative clones may become dominant after CAR T-cell therapy [72]. Previous exposure to the CAR T-cell therapy target may be another factor accounting for treatment failure because monoclonal antibodies targeting CD19 and BCMA have been widely used in patients with lymphoid malignancies. In B-cell ALL, most patients are treated with CD19-targeting treatments, such as blinatumomab, before CD19 CAR T-cell therapy. High response rates to CD19 CAR T-cell therapy were observed in patients with prior exposure to blinatumomab as long as tumor cells maintained CD19 expression [73, 74]. In contrast, nonresponders to blinatumomab had inferior outcomes to CD19 CAR T-cell therapy compared with the outcomes of patients who were either blinatumomab-naïve or had previously responded to blinatumomab [75]. Therefore, regardless of previous exposure to treatments targeting the same antigen, CAR T-cell therapy can be considered for patients retaining the target antigen. Besides antigen loss, the intrinsic nature of the tumor may be related to refractoriness to CAR T-cell therapy. When tumor cells harbor impaired death receptor signaling, such as the lack of proapoptotic molecules, they can resist CAR T-cell treatment [76].
Second, host factors can influence the cytotoxic function of CAR T cells and promote poor expansion and short persistence of infused CAR T cells, which may result in treatment failure. CAR T cells interact with tumor cells within the tumor microenvironment. The enrichment of negative factors, such as regulatory T cells, myeloid-derived suppressor cells, and immunosuppressive cytokines, including IL-4, IL-10, and transforming growth factor-β, can suppress CAR T-cell function [77]. In an evaluation of pretreatment tumor tissues and blood samples of DLBCL patients, tumor expression of interferon signaling and high levels of monocytic myeloid-derived suppressor cells, IL-6, and ferritin were associated with a lack of a durable response [78]. As CAR T cells are produced from autologous T cells of patients, T-cell fitness is another issue. Indeed, T cells from young donors have a higher transduction efficiency, and young donor-derived CAR T cells show better proliferation and cytotoxicity than those derived from older adults. Besides donor age, their physical fitness, number of previous treatments, tumor burden, and immunosuppressive features can influence T-cell fitness and CAR T-cell therapy outcomes [79]. Further, T-cell exhaustion hinders effective CAR T-cell function as exhausted T cells may arise from prior chemotherapy or alterations in the tumor microenvironment [80]. CAR T-cell exhaustion also can occur because of excessive antigenic signaling in the tumor environment, resulting in functionally impaired T cells characterized by the expression of checkpoint molecules, such as programmed cell death-1, T-cell immunoglobulin and mucin domain-containing protein-3, and lymphocyte activation gene-3, and decreased proliferation and secretion of effector cytokines.
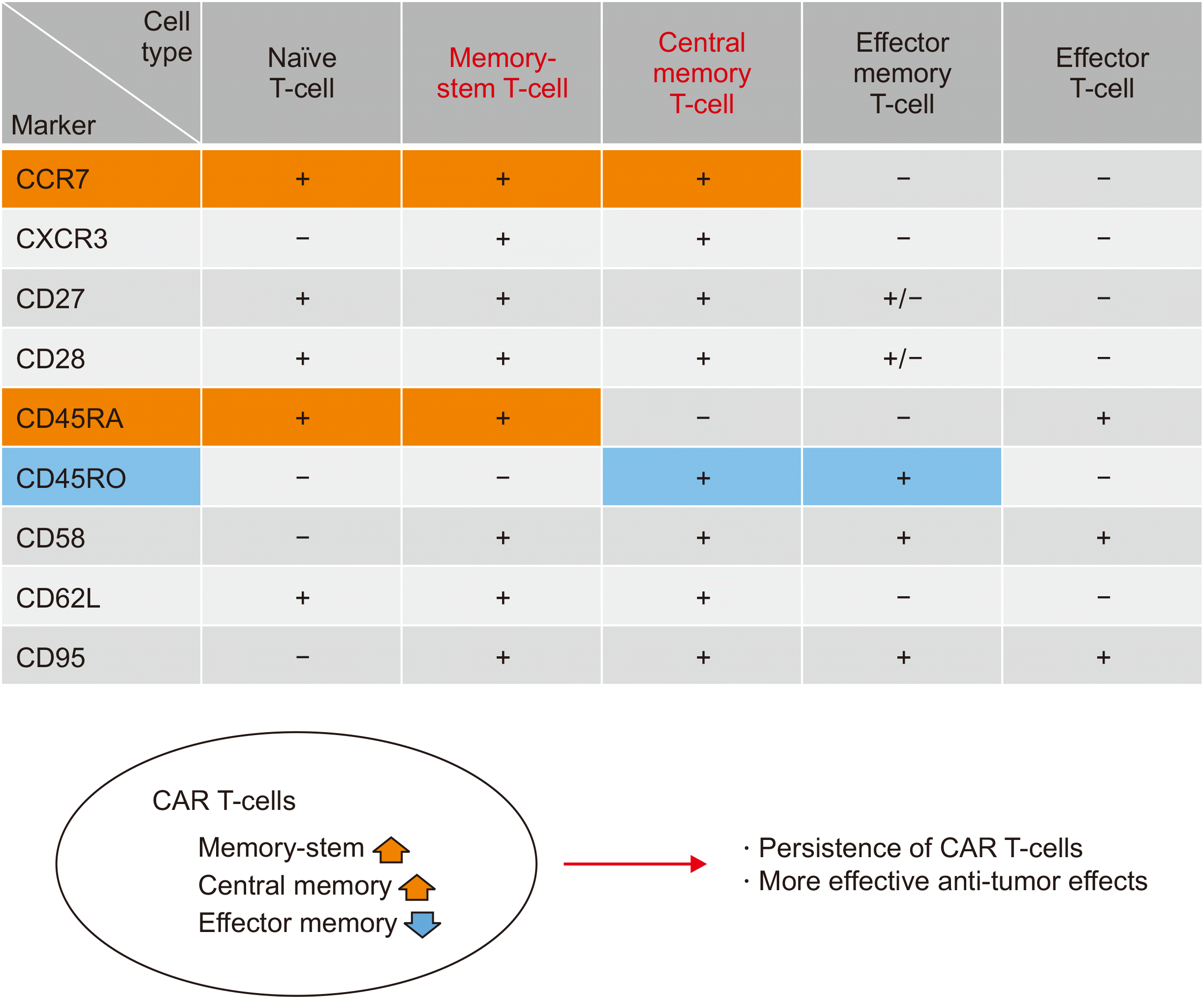
Third, the composition of the CAR T-cell product may influence the efficacy of CAR T-cell therapy because peripheral blood T-cell subsets can be quite heterogeneous in patients with B-cell lymphoid malignancies, and various factors, including age, disease subtype, and chemotherapy regimens before apheresis, can influence the proportions of naïve, memory, and effector T cells. During the differentiation of T cells, CD45RA+/CD45RO–/CCR7+ naïve T cells become CD45RO+/CCR7+ central memory and CD45RO+/CCR7– effector memory T cells and finally differentiate into effector T cells (Fig. 4). Single-cell RNA sequencing of the infused CAR T-cell products from patients receiving axi-cel for DLBCL revealed enrichment of CCR7+/CD27+ T cells in patients who achieved CR compared with that seen with cells from patients with PR/PD [81]. The enrichment of central memory T cells over effector memory T cells may account for the favorable outcomes of patients receiving CAR T cells. Likewise, an analysis of the CD8+ and CD4+ T-cell subsets in patients with B-cell malignancies showed a markedly increased proportion of effector memory T cells and a reduced proportion of naïve T cells compared with that in normal donors, implying an unfavorable composition of T cells [82]. Memory stem T cells are another rare subset of T cells bridging naïve T cells and conventional memory T cells, accounting for <3% of the entire T-cell population. Memory stem T cells are positive for CD45RA, CD62L, and CCR7 and negative for CD45RO, akin to naïve T cells, but they express CD95, CXCR3, and CD58, similar to central memory T cells [83]. Because memory stem T cells can self-renew and contribute to the generation of conventional memory and effector T cells, CAR T-cell products with a high proportion of memory stem T cells reportedly showed better antitumor effects than those with a low proportion in human lymphoma animal models [84, 85]. Likewise, an analysis of infused CAR T-cell products in the ZUMA-1 study showed that the number of CD45RA+/CCR7+ T cells was significantly related to durable responses, implying the role of CAR T-cell products enriched in naïve and memory stem T cells in achieving favorable outcomes [86]. Therefore, the proportion of memory stem T cells in CAR T-cell products may have a major impact on the efficacy of CAR T cells, and the development of CAR T-cell products enriched in memory stem T cells may enhance their clinical efficacy. A recent phase I/II study with memory-enriched CD19-directed CAR T cells demonstrated the favorable activity and safety of naïve and memory stem T-cell-derived CAR T cells in adult patients with r/r B-ALL [87].
Go to :
CONCLUSIONS
The introduction of CAR T-cell therapy in managing patients with r/r lymphoid malignancies has changed the treatment paradigm and provided tremendous benefits for patients with previously incurable diseases. However, challenging issues regarding antitumor efficacy and toxicities associated with treatment remain. Future research on ways to circumvent treatment failure and prevent adverse events after CAR T-cell therapy will enable us to improve patient outcomes. CAR T-cell therapy will become increasingly important in treating lymphoid malignancies.
Go to :
 XML Download
XML Download