

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
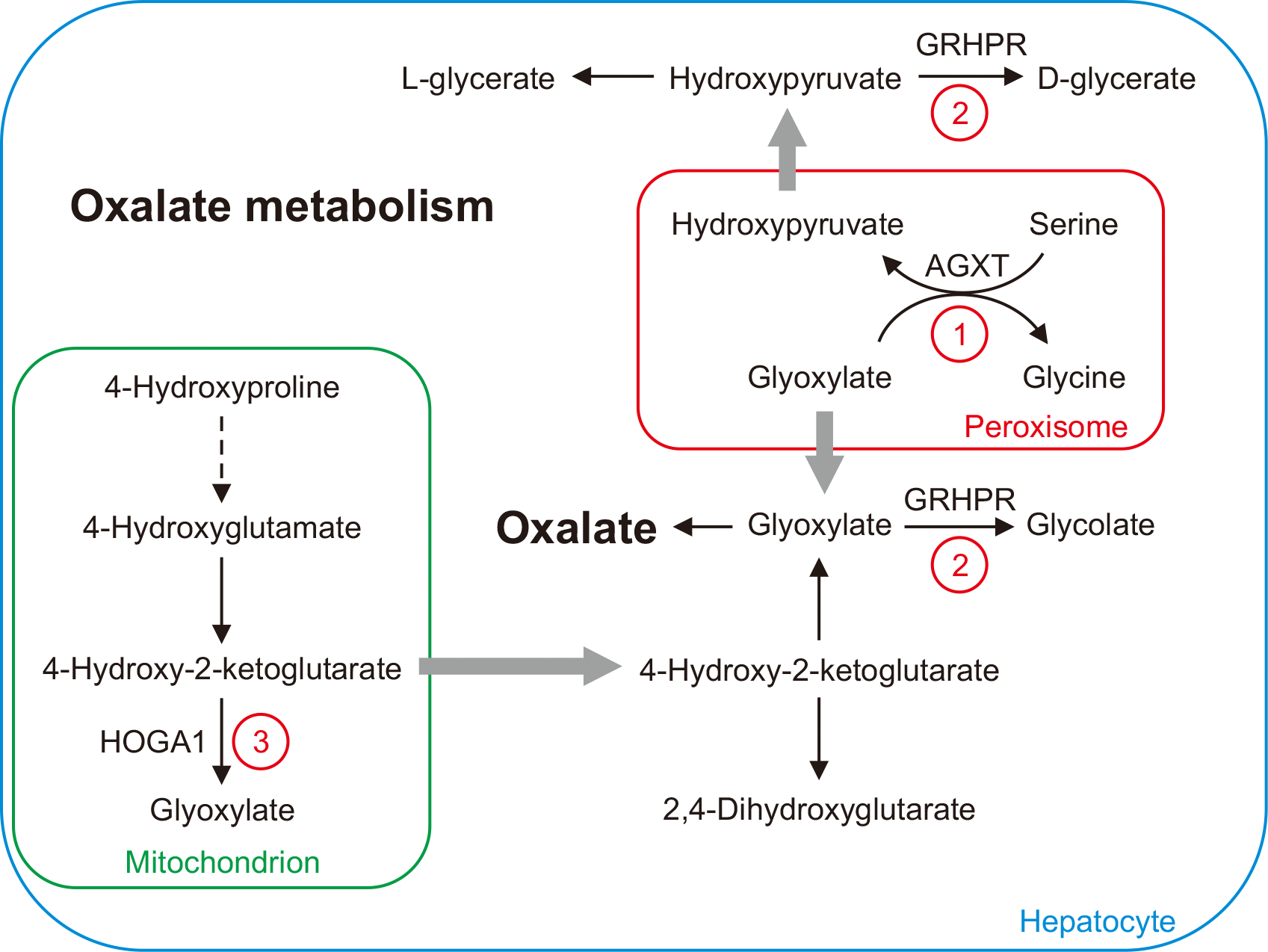
Endogenous oxalate is a metabolic end-product that is synthesized mainly in the liver and excreted in urine (Fig. 1) [1]. Primary hyperoxaluria (HP) types I, II, and III are inborn errors of metabolism associated with defects in oxalate metabolism. Primary HP, type I (HP1, OMIM #259900) is caused by a deficiency of liver-specific alanine-glyoxylate aminotransferase (AGXT) [2, 3]. HP1 is characterized by elevated plasma oxalate and urinary oxalate and glycolate concentrations [4]. Primary HP, type II (HP2, OMIM #260000) is caused by a deficiency of glyoxylate reductase/hydroxypyruvate reductase (GRHPR) and is characterized by elevated plasma oxalate and urinary oxalate and l-glycerate concentrations [5, 6]. Primary HP, type III (HP3, OMIM #613616) is due to a defect in 4-hydroxy-2-oxoglutarate aldolase 1 (HOGA1) and is characterized by elevated plasma oxalate and urinary oxalate, 4-hydroxyglutamate, and 2,4-dihydroxyglutarate concentrations [7, 8].
Quantitative measurements of oxalate concentrations in the urine and plasma are commonly used to screen, diagnose, and therapeutically monitor HP. Measuring urine oxalate concentrations is a common practice for suspected stone diseases and can be performed when measuring a panel of other metabolites that are elevated in hyperoxaluria. Plasma oxalate is particularly valuable for patients with renal failure or end-stage renal disease (ESRD) because urinary oxalate concentrations may be less reliable in these cases. In addition, patients undergoing hemodialysis require plasma oxalate measurements during monitoring tests. Establishing plasma oxalate testing is important for screening and therapeutically monitoring primary HP.
Common methods for plasma oxalate quantification include enzymatic assays, ion-exchange chromatography, and liquid chromatography-tandem mass spectrometry (MS) with solid-phase extraction [9-11]. Two different plasma oxalate measurement methods involving gas chromatography (GC)-MS were reported in 1973 and 1988. Both methods require large sample volumes (5 and 4 mL of plasma, respectively) and have unsatisfactory recovery rates (69%±4% and 57.9%, respectively) [12, 13]. A GC-MS method for quantifying plasma oxalate involving N,O-bis(trimethylsilyl)trifluoroacetamide (BSTFA) derivatization was reported in 2003; however, the trimethylsilyl derivative caused co-elution interference on a DB-5MS column, as reflected by an unknown peak at m/z 219 [14]. This method requires 2 mL of plasma, which is greater than the specimen volume required for enzymatic assays [14, 15].
We developed a sensitive GC-MS procedure for oxalate quantitation that requires only 250 μL of plasma.
MATERIALS AND METHODS
The experiments involving human subjects were conducted in accordance with the principles outlined in the Declaration of Helsinki. This study was approved by the Research Ethics Board (REB) of the Hospital for Sick Children (REB No.: 1000081081).
Materials
Oxalate, 5-sulfosalicylic acid, N-tert-butyldimethylsilyl-N-methyltrifluoroacetamide with 1% tert-butyldimethylchlorosilane (MTBSTFA+1% TBDMS), and human Hb were purchased from Sigma-Aldrich (St. Louis, MO, USA). Oxalate-1,2-13C2 was purchased from Cambridge Isotope Laboratories (Andover, MA, USA), and an SPB-1 capillary column (30 m×0.25 mm×0.25 mm film thickness) was purchased from Supelco (Bellefonte, PA, USA). Ethyl acetate and hexane were purchased from EMD Chemicals (Gibbstown, NJ, USA). All other chemicals were of analytical grade.
Preparation of standard solutions and QC materials
Stock solutions of oxalate (10 mM) and 13C2-oxalate (10 mM) were prepared in H2O. A working solution of the internal standard (IS; 400.0 μmol/L) was prepared by diluting a 13C2-oxalate stock solution with H2O. A calibrator set (calibrator A–H) with eight oxalate concentrations (0.0, 1.0, 8.9, 16.8, 32.6, 48.4, 64.2, and 80.0 μmol/L) was prepared in saline solution (0.9% NaCl). Specifically, calibrator A (0.0 μL) was saline solution. Calibrators B (1.0 μmol/L) and H (80.0 μmol/L) were prepared by adding 1 or 80 μL of oxalate stock solution (10.0 mmol/L), respectively, to 10 mL of saline solution. The other calibrators were prepared by adding various volumes of calibrators B or H to the saline solution.
Normal QC material was prepared by pooling and acidifying heparin plasma freshly collected from normal subjects. Spiked samples were prepared by adding oxalate solution to the normal QC material before processing, resulting in final oxalate concentrations of 2.0, 5.0, 10.0, 20.0, and 40.0 μmol/L.
Blood collection
Overnight fasting whole blood specimens were collected in lithium heparin Vacutainer tubes and immediately placed on ice. Plasma was separated within 30 mins of collection by centrifugation at 2,000×g at 4°C. The plasma pH was adjusted to <2.0 by adding ~20 μL of concentrated HCl/mL of plasma and the acidified plasma was stored at –20°C.
A total of 330 pediatric specimens were included in the cut-off analysis: 65% of the specimens were from patients newly diagnosed as having HP1 or HP3 (and undergoing monitoring), 8% were from patients with renal stones, and 21% were from patients with other renal disorders (such as acute kidney injury [AKI], disorders requiring a kidney transplant, renal failure, renal dysplasia, chronic kidney disease [CKD], ESRD, nephronophthisis, echogenic kidneys, neutropenia, hematuria, and renal scarring). The remaining specimens (5%) were obtained from patients with other conditions (such as autism, chest pain, epilepsy, familial hypomagnesemia with hypercalciuria and nephrocalcinosis, global developmental delay, multiple anomalies, hereditary paragangioma syndrome, hyperkalemia, hypertension, Kabuki syndrome, subdural hematomas, and VACTERL association) or patients without clinical information (1%).
Sample preparation
Two hundred fifty microliters of each plasma sample was spiked with 50 μL of IS (400.0 mol/L oxalate-1,2-13C2) and vortexed for 2 secs. Then, 250 μL of 5-sulfosalicylic acid (10%) was added, and the sample was vortexed for 10 secs. The samples were centrifuged at 15,600×g for 5 mins to remove proteins, and the supernatants were transferred to clean borosilicate tubes. NaCl (0.5 g) and 200 μL of HCl (2N) were added to each tube, and the contents were vortex-mixed for 5 secs. Oxalate was extracted twice with 2 mL of ethyl acetate. The two extracts were combined and evaporated to dryness under a stream of nitrogen gas at 40°C. Oxalate was derivatized with 50 μL of MTBSTFA+1% TBDMS at 100°C for 1 hr. Finally, 50 μL of hexane was added to each sample and an aliquot (1 μL) of each sample was analyzed by GC-MS.
GC-MS analysis
The GC injector temperature was set to 280°C, and the initial column temperature was set to 60°C. The temperature was increased to 180°C at 10°C/min and then to 310°C at 25°C/min. The temperature was maintained at 310°C for 4 mins. The GC-to-MS interface temperature was set to 280°C. The mass spectrometer was run in positive-electron impact mode with an energy of –70 eV and a source temperature of 260°C. Prior to each analytical run, the GC column compartment temperature was maintained at 300°C for a minimum of 1 hr. Before each analytical run, the column was conditioned with two injections of MTBSTFA and 1% TBDMS. Samples were analyzed in splitless mode using a 5975 GC-MS instrument (Agilent, Santa Clara, CA, USA). Oxalate was eluted from an SPB-1 capillary column (30 m×0.25 mm ID×0.25 mm) using helium (purity: 99.999%) as the carrier gas at a flow rate of 1 mL/min. Selected ion-monitoring mode at m/z 261.10 was used to detect oxalate and at m/z 263.15 to detect oxalate-1,2-13C2 (Supplemental Data Table S1). A MassHunter workstation (Agilent) was used to acquire and process the data.
Method validation
Assay linearity was assessed by analyzing eight calibrators with oxalate concentrations of 0.0, 1.0, 8.9, 16.8, 32.6, 48.4, 64.2, and 80.0 μmol/L. Each calibrator was measured in triplicate. The acceptance criteria for this validation included ensuring linearity across the calibration range, no obvious outliers, and a high R2 value (≥0.98). The upper limit of quantification was the inflection point in the plot.
The limit of blank (LoB) was determined by measuring oxalate in four aliquots of a blank sample, using the following equation:
LoB=mean oxalate concentration of the blank sample+1.65 SD of the blank sample
The limit of detection (LoD) was determined by measuring oxalate in four aliquots of pooled patient specimens diluted to low concentrations. The aliquots were measured in triplicate for three days. The LoD was calculated using the following equation:
LoD=oxalate LoB+1.65 SD of the low-concentration sample
The lower limit of quantification (LLoQ) was determined using four serially diluted samples. A QC sample with a low oxalate concentration was serially diluted with saline solution so that, at minimum, four dilutions extended below calibrator B (5.0 μmol/L) and approached calibrator A (0.0 μmol/L). All samples were analyzed in triplicate on the same day. The expected oxalate concentrations were 5.0, 2.5, 1.25, and 0.625 μmol/L for the neat and 2×, 4×, and 8× dilutions, respectively. The acceptance criterion was that the LLoQ equaled the lowest concentration with a %CV of <20% or the LoD, whichever was higher.
The between-day precision was assessed using pooled plasma samples with three different oxalate concentrations. Each concentration was measured five times per day for five days (N=25 replicates). The acceptance criteria were the low-concentration QC sample having a %CV of ±30% and the high- and medium-concentration QC samples having a %CV of ±10%.
Recovery was determined by analyzing spiked samples four times. The average concentration in each spiked sample is the total endogenous oxalate concentration in the normal QC material plus the spiked oxalate concentration. Therefore, the measured concentration of spiked oxalate was calculated by subtracting the endogenous oxalate concentration from the total concentration. Recovery was calculated using the measured and targeted concentrations of spiked oxalate. The acceptance criterion for recovery was a value within 100%±20%.
Carryover was assessed by conducting successive measurements of three aliquots (a1, a2, and a3) of a plasma sample containing a high oxalate concentration (65.3 μmol/L), followed by successive measurements of three aliquots (b1, b2, and b3) of plasma containing a low oxalate concentration (2.7 μmol/L). Carryover (k) was calculated using the following equation:
k=(b1–b3)/(a3–b3)
Carryover was validated by analyzing a plasma sample with a high oxalate concentration, followed by analyzing the hexane/MTBSTFA mix. The acceptance criterion for carryover was a value ≤1.0%.
Interference due to hemolysis, lipemia, and icterus was assessed by analyzing plasma samples spiked with human Hb (5.0 g/L), intralipids (17.0 mmol/L), and total bilirubin (500.0 mol/L), respectively. Neat and spiked samples were measured in quadruplicate.
The effect of pH on oxalate concentrations was assessed using acidified heparin, non-acidified heparin, and non-acidified EDTA plasma. Immediately after collection, each specimen was aliquoted into two tubes: one with EDTA preservative and the other with lithium heparin. All tubes were placed in an ice-water bath until the plasma was separated. A heparinized plasma aliquot from each sample was further divided into two aliquots: one aliquot was acidified with concentrated HCl to a pH of <2.0 and the other was left untreated. The EDTA plasma aliquots were not acidified. The oxalate concentrations of 15 samples were measured.
The dilution effect was evaluated using a specimen with an oxalate concentration of ~80.0 µmol/L. The specimen was aliquoted and diluted 2×, 5×, and 10× in 5% bovine serum albumin solution. The oxalate concentrations in neat and diluted aliquots were measured in triplicate. The acceptance criterion was the normalized concentrations being within 10% of those in undiluted specimens.
Method comparison
Method comparison was conducted by performing GC-MS and enzymatic assays. Specimens from 12 patients with oxalate concentrations ranging between 0.0 and 80.0 mol/L were measured using the GC-MS method. Aliquots of these specimens were sent to an external laboratory for testing using an enzymatic assay based on oxalate reduction by oxalate oxidase. The reaction releases hydrogen peroxide, which in the presence of peroxidase reacts with a dye to give a colored endpoint that can be measured at 590 nm [16]. The results were analyzed using linear regression and the differences between both methods plotted. The acceptance criteria were concentrations <5.0 μmol/L demonstrating a bias within ±1.5 μmol/L and concentrations above 5.0 μmol/L demonstrating a bias within ±30%.
Cut-off analysis
Three hundred thirty results from pediatric patients were retrospectively analyzed using the GC-MS method. Of these 330 results, 215 were from 11 patients with HP (seven with HP1 and four with HP3) and 115 were from 78 patients without HP who had various kidney disorders. The results from patients with HP before treatment were plotted against those from patients without HP. ROC curves were plotted, and the area under the ROC curve (AUC) value, optimal cut-off, specificity, and sensitivity were calculated. Considering that this method can be used as a screening test for HP, the cut-off with 100% specificity was also analyzed.
Statistical analysis
Statistical analysis was performed using Microsoft Excel (Microsoft, Seattle, WA, USA). ROC curves and boxplots were drawn using MetaboAnalyst 5.0 (Montreal, Canada) [17].
RESULTS
Chromatography
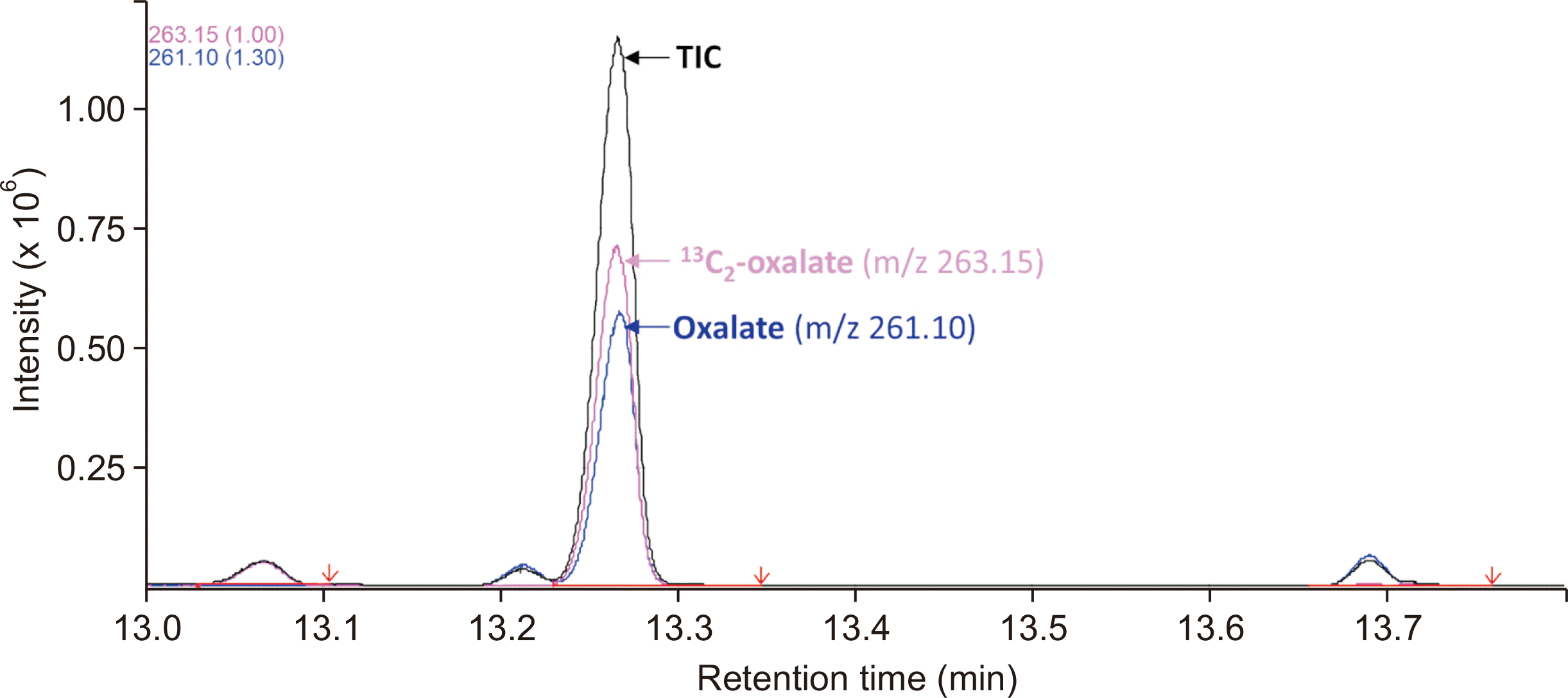
The GC-MS total ion chromatograph of plasma oxalate displayed a peak eluting at 13.26 mins, which included both oxalate (m/z: 261.1) and the IS (13C2-oxalate; m/z: 263.15; Fig. 2). The oxalate peak was baseline-resolved from those of other closely eluting molecules. We noticed that analyzing plasma oxalate without pre-conditioning the GC column could lead to a false elevation of the oxalate peak. Maintaining the GC column at 300°C for 1 hr before each analytical run prevented this artifact.
Method validation
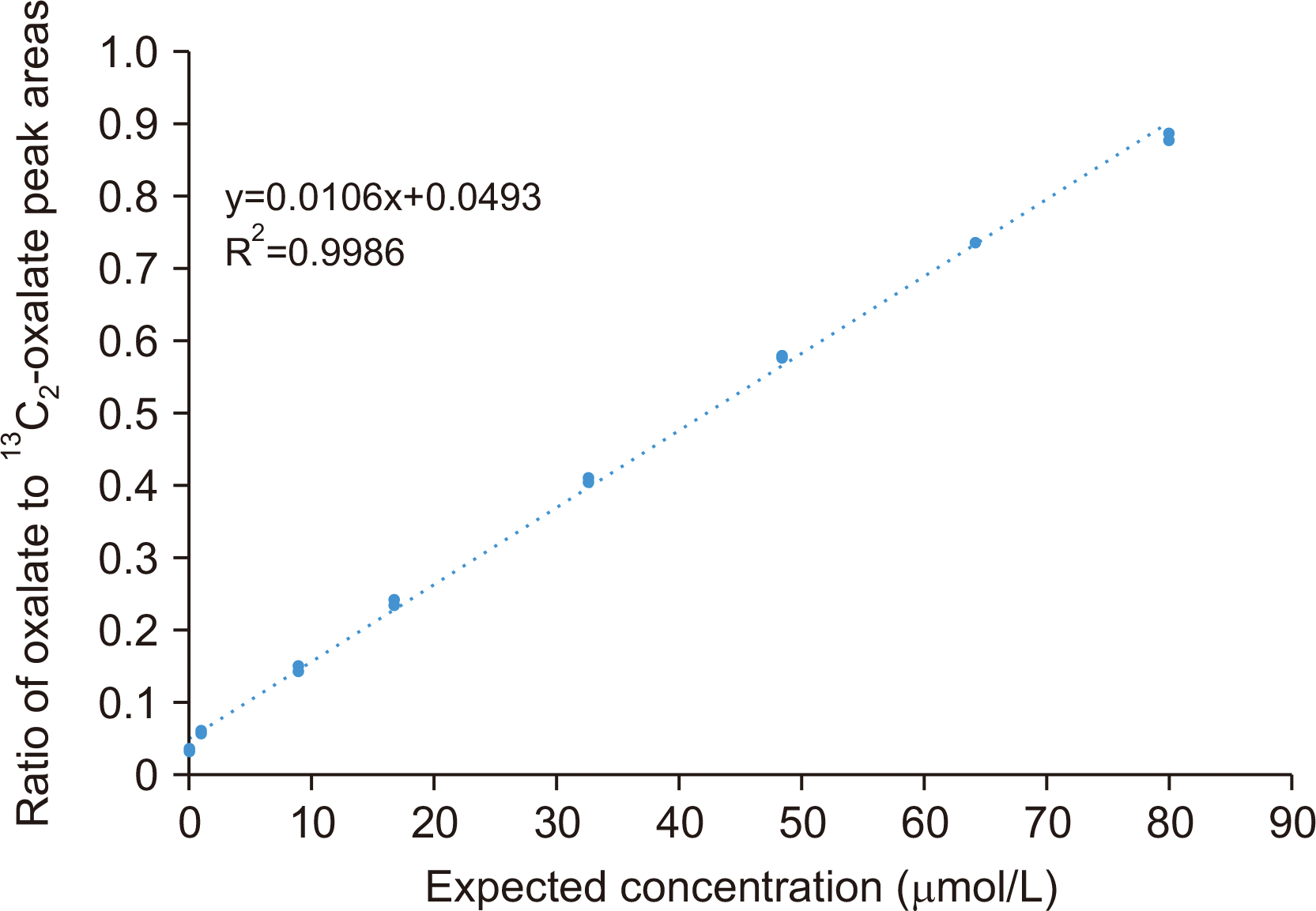
Linearity was assessed by analyzing eight calibrators with measured oxalate concentrations of 1.3, 3.6, 11.6, 19.9, 35.4, 50.9, 65.4, and 79.5 μmol/L. Triplicate data for calibrators A–H showed CVs of 13%, 3%, 2%, 2%, 0%, 0%, 0%, and 1%, respectively. The linearity analysis showed that the assay was linear up to 80.0 μmol oxalate/L, with an R2 value of 0.9986, a slope of 0.0106, and an intercept of 0.0493 (Fig. 3). The standard errors of the slope and intercept were 0.000088 and 0.003459, respectively. The LoB and LoD were 0.24 and 0.78 μmol/L, respectively. The lowest oxalate concentration among the diluted QC samples with a %CV <20% was 0.6 μmol/L (16% CV), which was lower than the LoD. The LLoQ was determined to be 0.78 μmol/L, and the linear range of the assay was 0.8 (rounded up from 0.78) to 80.0 μmol/L.
Precision analysis showed that the %CV was 16% at a low oxalate concentration of 3.2 μmol/L, 2% at a medium concentration of 36.4 μmol/L, and 1% at a high concentration of 65.8 μmol/L (Supplemental Data Table S2).
The recovery was between 90% and 110% when the concentration of spiked oxalate ranged from 2.0 to 40.0 mol/L (Supplemental Data Table S3).
The carryover was 0.16% when a plasma sample with a low oxalate concentration was run immediately after a sample with a high oxalate concentration. Analysis of the hexane/MTBSTFA mix following plasma samples did not reveal a detectable signal within the retention-time window for oxalate.
Interference analysis showed that Hb did not interfere with the results. However, triglycerides and bilirubin led to falsely elevated oxalate concentrations. Triglycerides (17.0 mmol/L) in plasma with 1.2 μmol/L oxalate caused a 2.5-fold increase in false positives. Adding 500 µM bilirubin to the plasma samples increased the false-positive rate by 158% (Supplemental Data Table S4).
Oxalate concentrations were markedly higher when the plasma was not acidified. As shown in Supplemental Data Table S5, oxalate concentrations in non-acidified heparin and EDTA plasma samples were 3–7-fold higher than those in acidified heparin plasma samples. No significant difference in oxalate concentrations was found between non-acidified heparin and non-acidified EDTA plasma samples (P=0.48).
Dilution effect analysis showed that the percentage differences between neat and diluted samples were all within 6% (5.5% for the 2× dilution, 5.9% for the 5× dilution, and –5.1% for the 10× dilution), indicating that the dilution effect was minimal.
The minimum required sample volume was 250 μL. This volume was determined considering the requirements of the assay procedure, GC-MS sensitivity, sample volume availability, and ease of sample collection in the pediatric population.
Method comparison
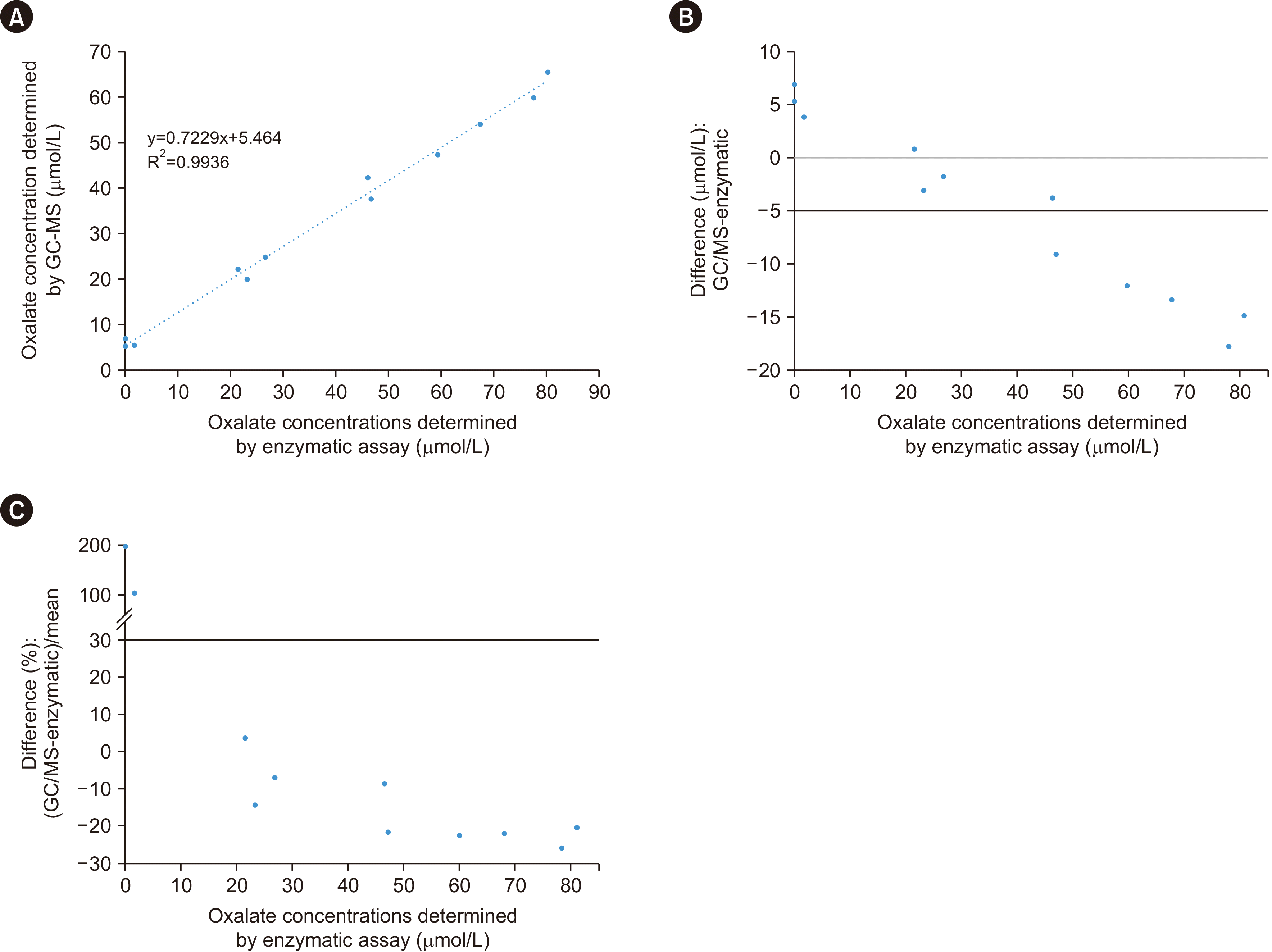
Linear regression analysis of two sets of measurements revealed a linear relationship, with an R2 value of 0.9936 (Fig. 4A). However, difference plots indicated that the enzymatic assay could not be used to detect oxalate in specimens with low oxalate concentrations (5.3 or 6.9 μmol/L, as measured by GC-MS) (Fig. 4B and 4C). The enzymatic activity in the specimens (1.7 μmol/L) was significantly lower than that measured using GC-MS (5.5 μmol/L). At concentrations >47.0 mol/L, the enzymatic assay results were consistently higher than those obtained by GC-MS, with an average difference of 20%, although this bias is unlikely to be clinically relevant (Fig. 4B and 4C; Supplemental Data Table S6).
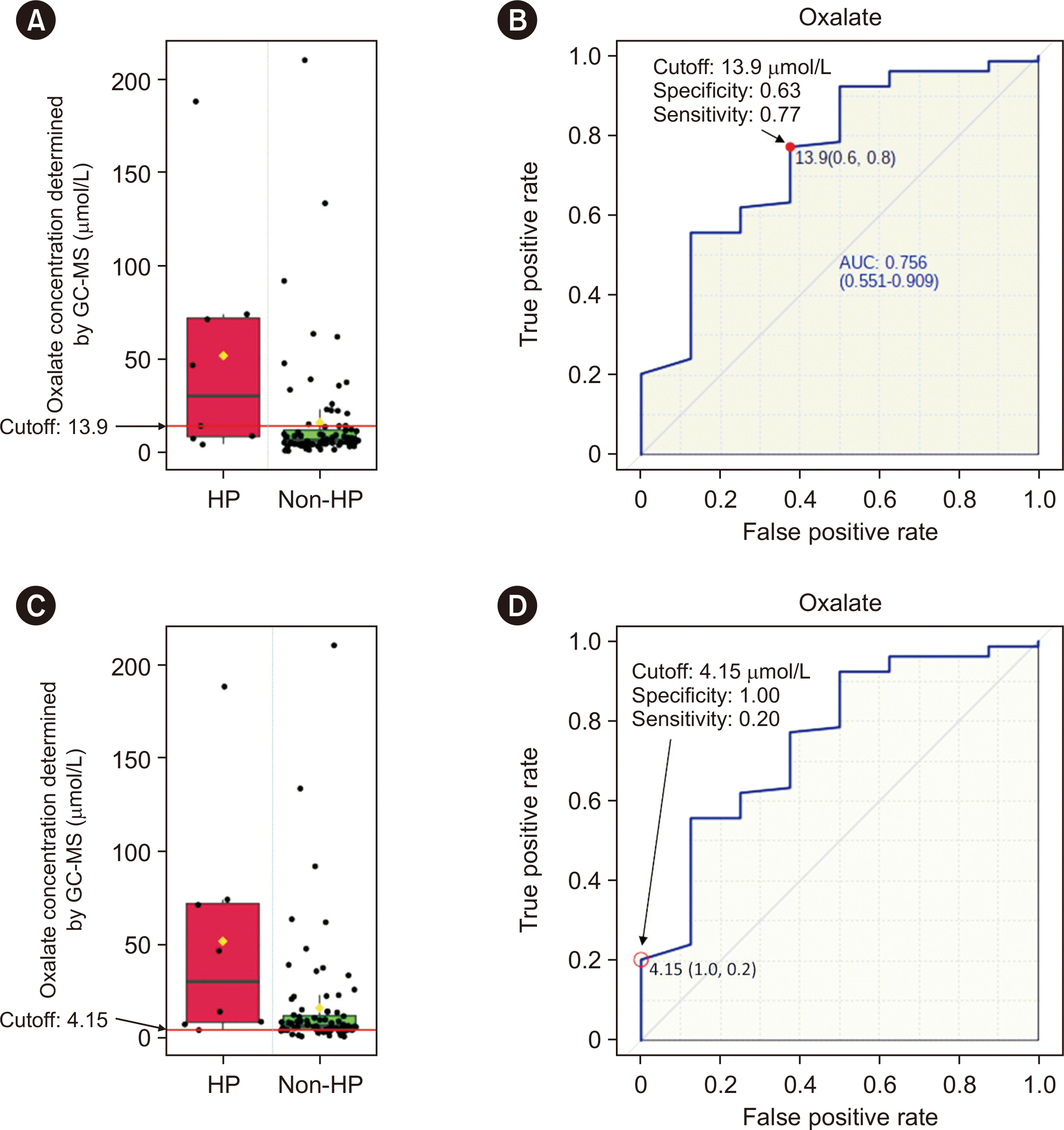
Cut-off analysis
Patients with or without HP demonstrated a wide range of plasma oxalate concentrations. For example, in patients with HP before treatment, the lowest oxalate concentration was 4.2 μmol/L (in a patient with HP3) and the highest concentration was 187.9 μmol/L (in a patient with HP1) (Fig. 5A and 5C; data for patients with HP after treatment are not shown). The ROC curve indicated that the optimal cut-off for distinguishing between patients with or without HP was 13.9 μmol/L, with 63% specificity and 77% sensitivity (Fig. 5B). The AUC value was 0.756. A cut-off of 4.15 μmol/L showed 100% specificity and 20% sensitivity (Fig. 5D).
DISCUSSION
We developed a sensitive GC-MS procedure for quantifying plasma oxalate, with a detection limit of 0.78 μmol/L. This assay requires only 250 μL of plasma, making it convenient for use in pediatric patients with limited blood collection capacity. Unlike a previously reported GC-MS procedure [14], our method is not influenced by interference from trimethylsilyl derivatization. However, we observed unknown interference that caused falsely increased oxalate measurements at low concentrations. The origin of the interference remains unidentified, but it seems to have been caused by materials deposited in the GC column, specifically in the front-end section of the column that connects to the GC inlet. Maintaining the column temperature at 300°C for 1 hr effectively eliminated these materials. We sought to minimize matrix effects by using an isotope-labeled IS for instrument calibration, conducting analysis in plasma matrix where possible, and removing interfering proteins by salting them out and precipitating them with organic solvents [18, 19].
The results of the method comparison suggest that enzymatic assays may not be as sensitive as GC-MS. At oxalate concentrations <7.0 μmol/L, the enzymatic assay either yielded significantly lower results or was unable to detect oxalate. At oxalate concentrations >47.0 mol/L, the enzymatic assay results were consistently higher than those obtained by GC-MS, with an average difference of 20%. These findings contradict those of previously reported method comparisons, in which enzymatic assays yielded lower oxalate concentrations than GC-MS [20, 21]. Because of the unavailability of external QC samples, it is challenging to determine which method is more accurate for this analysis. A well-designed study should be conducted to further address this discrepancy.
Elevated plasma oxalate concentrations can stem from various causes, including HP and other conditions [22]. The metabolism of ascorbic acid (vitamin C) can contribute to increased plasma and urine oxalate concentrations [23-25]. The oxidation of ascorbic acid to dehydroascorbic acid (the first step toward oxalate synthesis) is pH-dependent [26]. To minimize the in vitro conversion of ascorbic acid to oxalate, specimen handling and management should be optimized, including immediate plasma separation, adjusting the pH to ≤2, and storing the acidified plasma at –20°C [27, 28]. Our results indicate that both green-top (heparin) and lavender-top (EDTA) tubes can be used for plasma collection, without significant differences in oxalate measurements.
Our findings indicate that intralipid and bilirubin concentrations can lead to false elevations in oxalate concentrations. We hypothesize that triglycerides and bilirubin may generate co-eluting fragment molecules with m/z ratios similar to that of oxalate (261.1). However, identifying these fragments is time-consuming and requires various technologies, such as time-of-flight-tandem MS, nuclear magnetic resonance, spectrophotometry, and plasma fractionation, some of which are unavailable in our laboratory. Our findings suggest that clinical conditions such as hyperbilirubinemia, intralipid infusion, and a high-fat diet may interfere with the test results. A retrospective analysis of patient results revealed elevated oxalate concentrations in clinical conditions associated with kidney dysfunction, including CKD, nephrocalcinosis, and AKI.
Various normal reference intervals for plasma oxalate concentrations have been reported [11]. The reference intervals obtained in different laboratories or with different methods vary over a relatively wide range. Establishing accurate normal reference ranges for plasma oxalate concentrations is challenging because of various factors and interferences, including specimen handling (e.g., fasting requirement, acidification, processing time, and storage conditions), diet, and potential clinical conditions. We recommend establishing an appropriate cut-off between patients with primary and patients with non-primary HP instead of establishing a normal reference range for the healthy population.
Our data obtained using specimens from a pediatric population showed that a cut-off of 4.15 μmol/L with 100% specificity can be used to screen for HP with a low chance of missing true-positive cases. Given its sensitivity of only 20%, it is important to correlate the results with clinical conditions, repeat plasma oxalate testing, and obtain a urinary panel test for proper follow-up. We propose an optimal cut-off of 13.9 μmol/L, with 63% specificity and 77% sensitivity.
This study had some limitations. Data from patients with HP2 were not included because such patients are currently not treated in our hospital. The suggested cut-offs should be further evaluated when more data from pediatric patients become available. In addition, the AUC value of 0.756 indicated that plasma oxalate testing alone is not appropriate for diagnosing HP. Instead, diagnosis should rely on abnormal patterns in urine (i.e., elevated glycolate, l-glycerate, and 4-hydroxyglutamate concentrations in patients with HP1, HP2, and HP3, respectively, along with an elevated oxalate concentration) and pathogenic variants identified by molecular genetic analysis. Another limitation of our study is that we used non-standard reference oxalate for method validation. However, the standard used was of analytical grade, which is sufficient for our clinical requirements and helped minimize our investment. Linearity analysis revealed accuracy only up to 80.0 μmol/L, which will be exceeded in patients with HP. For results that fall above the linearity range or for patients known to have HP, a diluted patient sample can be analyzed. Urinary oxalate concentrations are less reliable in patients with kidney dysfunction or ESRD [29]. In these cases, plasma testing is appropriate. In addition, patients undergoing hemodialysis require plasma oxalate measurements as a monitoring test [30]. Other secondary causes of hyperoxaluria include enteric hyperoxaluria, excessive ascorbic acid intake, and excessive dietary oxalate consumption.
In conclusion, we developed a highly sensitive GC-MS assay for plasma oxalate measurements that is particularly suitable for pediatric patients because of the low specimen volume required. The clinical context should be considered when interpreting plasma oxalate results.
SUPPLEMENTARY MATERIALS
Supplementary materials can be found via https://doi.org/10.3343/alm.2023.0178
 XML Download
XML Download