

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Posttransplant lymphoproliferative disorder (PTLD) represents a rare and severe complication of solid organ transplantation [1]. The incidence rates of PTLD after kidney transplantation (KT) range from 0.9 to 4.6 cases per 1,000 person-years [2]. Most PTLD cases originate from B cells infected with Epstein-Barr virus (EBV); consequently, management strategies predominantly target B cell-driven PTLD (B-PTLD). In contrast, T cell-driven PTLD (T-PTLD) occurs less frequently after solid organ transplantation [3]. The first-line treatment for PTLD involves the reduction of maintenance immunosuppression. However, no treatment options have been established for T-PTLD that is resistant to decreased immunosuppression [4]. Successful remission of refractory T-PTLD has been documented in only a limited number of studies, primarily case series and reports. Despite these efforts, the mortality rate associated with T-PTLD remains extremely high [5].
In this report, we present a fatal case of EBV-associated T-PTLD that rapidly progressed to acute liver failure, septic shock, and death. Despite various therapeutic interventions—including reduction of immunosuppression, introduction of a mammalian target of rapamycin inhibitor, administration of rituximab, and cytotoxic chemotherapy—treatment proved ineffective.
CASE REPORT
The study adhered to the principles of the Declaration of Helsinki and was approved by the Seoul National University Hospital Institutional Review Board (IRB No. H-2208-158-1354), which waived the requirement for patient consent. Written informed consent for the publication of this case report and any accompanying images was secured from the patient’s daughter, as the patient could not consent due to death.
A 50-year-old woman received a preemptive living donor KT from her daughter for diabetic end-stage kidney disease. She did not display preformed anti-human leukocyte antigen antibodies, and her blood type was compatible with the donor type. The patient received inductive basiliximab and a maintenance triple immunosuppressive regimen consisting of tacrolimus, mycophenolate mofetil (MMF), and prednisolone. Table 1 presents the serologic profiles of the donor and recipient before and after the transplant. EBV viral capsid antigen immunoglobulin G antibodies (>750 U/mL) were detected in both donor and recipient prior to KT. Because the recipient’s kidney function remained stable without evidence of rejection, we halved the tacrolimus dosage, discontinued MMF, and initiated sirolimus at a dosage of 1 mg daily, leveraging its antiviral properties. Nevertheless, the EBV viral load approximately doubled, reaching 652,111 copies/mL 4 months after transplantation. Consequently, we administered preemptive rituximab at a dose of 375 mg/m2 for 6 months after KT.
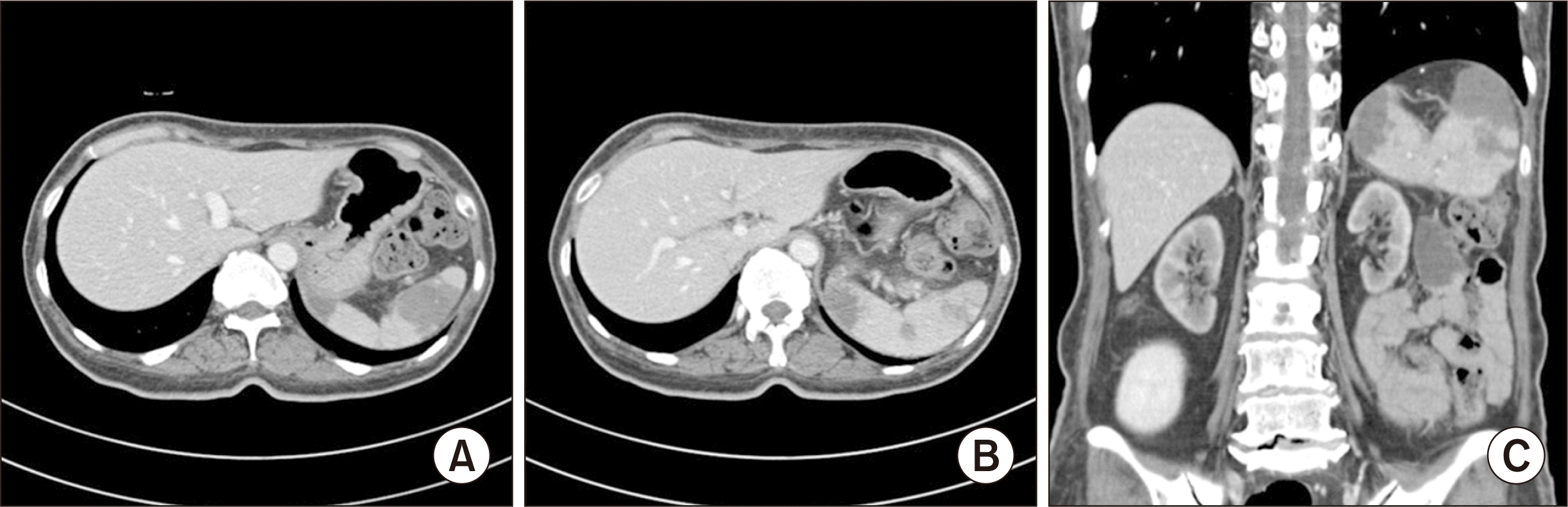
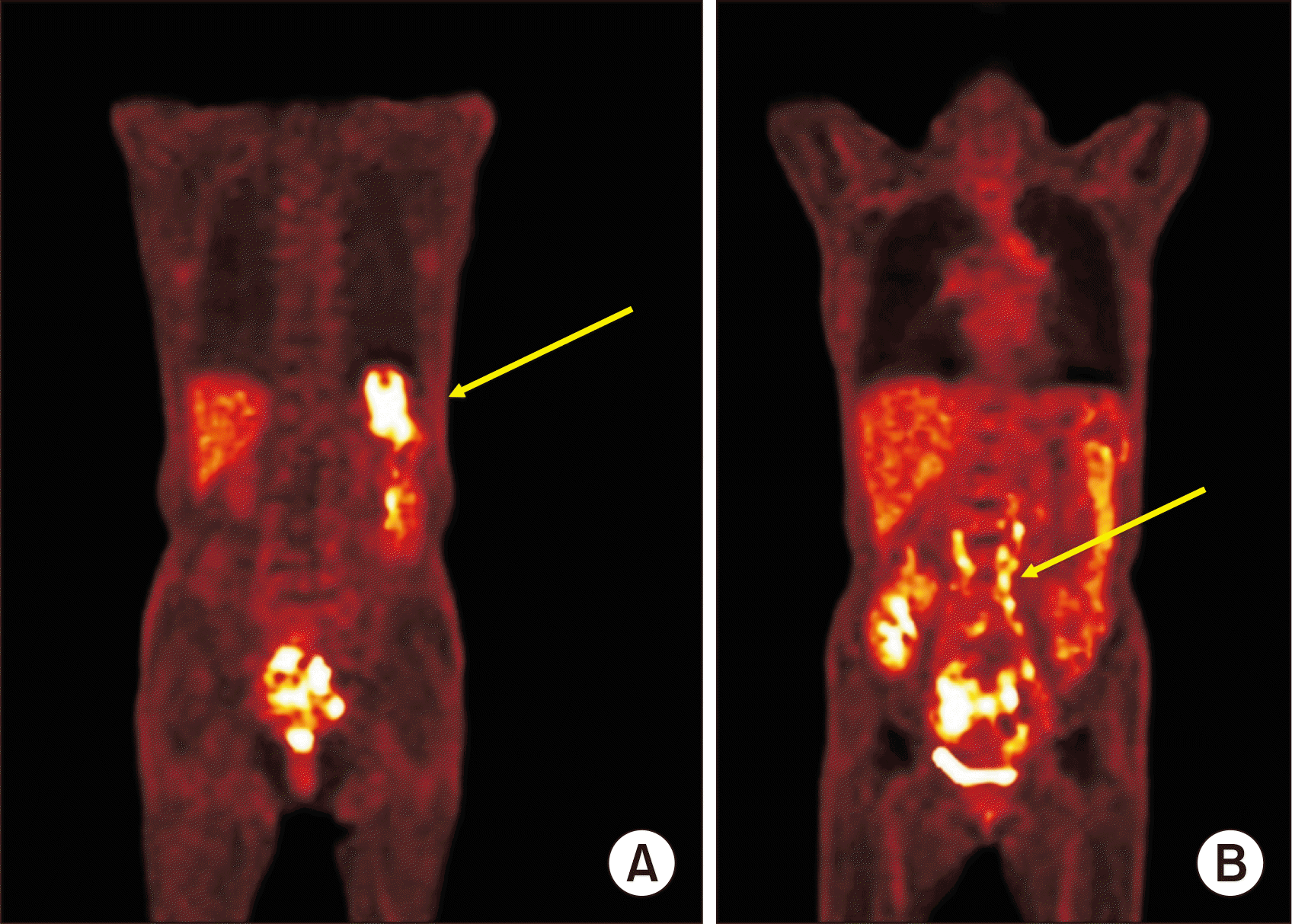
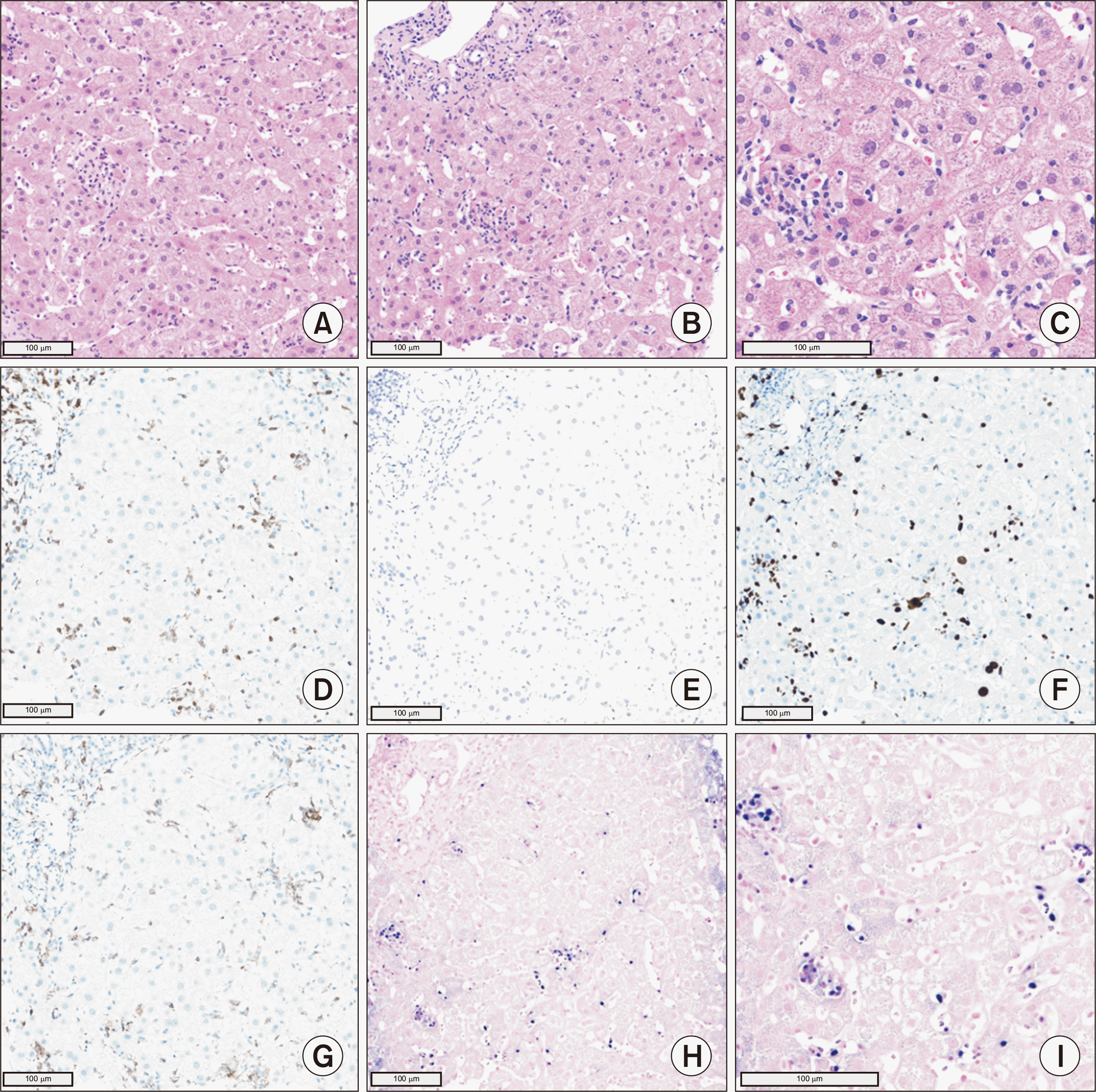
Eight months following KT, the patient’s EBV load had decreased to 375,412 copies/mL. However, new multifocal wedge-shaped lesions with low enhancement were detected on abdominopelvic computed tomography (APCT), prompting suspicion of splenic infarction (Fig. 1). Attempts to determine the cause of the infarction using peripheral blood smear, echocardiography, and coagulation-related blood tests were unsuccessful. Nine months after KT, the patient was admitted to the hospital with a fever of 40 °C, flank pain, abnormal liver function (as evidenced by an alanine transaminase [ALT] level of 161 IU/L), and an elevated EBV load of 1,921,952 copies/mL. APCT revealed para-aortic lymph node enlargement and increased splenic lesion margins. Because the clinical presentation was suggestive of PTLD, we performed positron emission tomography (PET)/CT, which revealed heightened metabolic activity in the spleen and lymph nodes along both iliac chains (Fig. 2). A liver biopsy, conducted under suspicion of PTLD involvement, confirmed EBV-associated PTLD of the T cell lineage, with expression of CD3 and CD56 (Fig. 3). The neoplastic lymphoid cells tested positive for CD3, CD4, CD8, CD56, Ki-67, granzyme B, and EBV in situ hybridization, but they were negative for CD20. In the absence of an established treatment for T-PTLD, we opted to further reduce immunosuppression. The patient continued on tacrolimus, oral prednisolone, and sirolimus, while we monitored her condition closely.
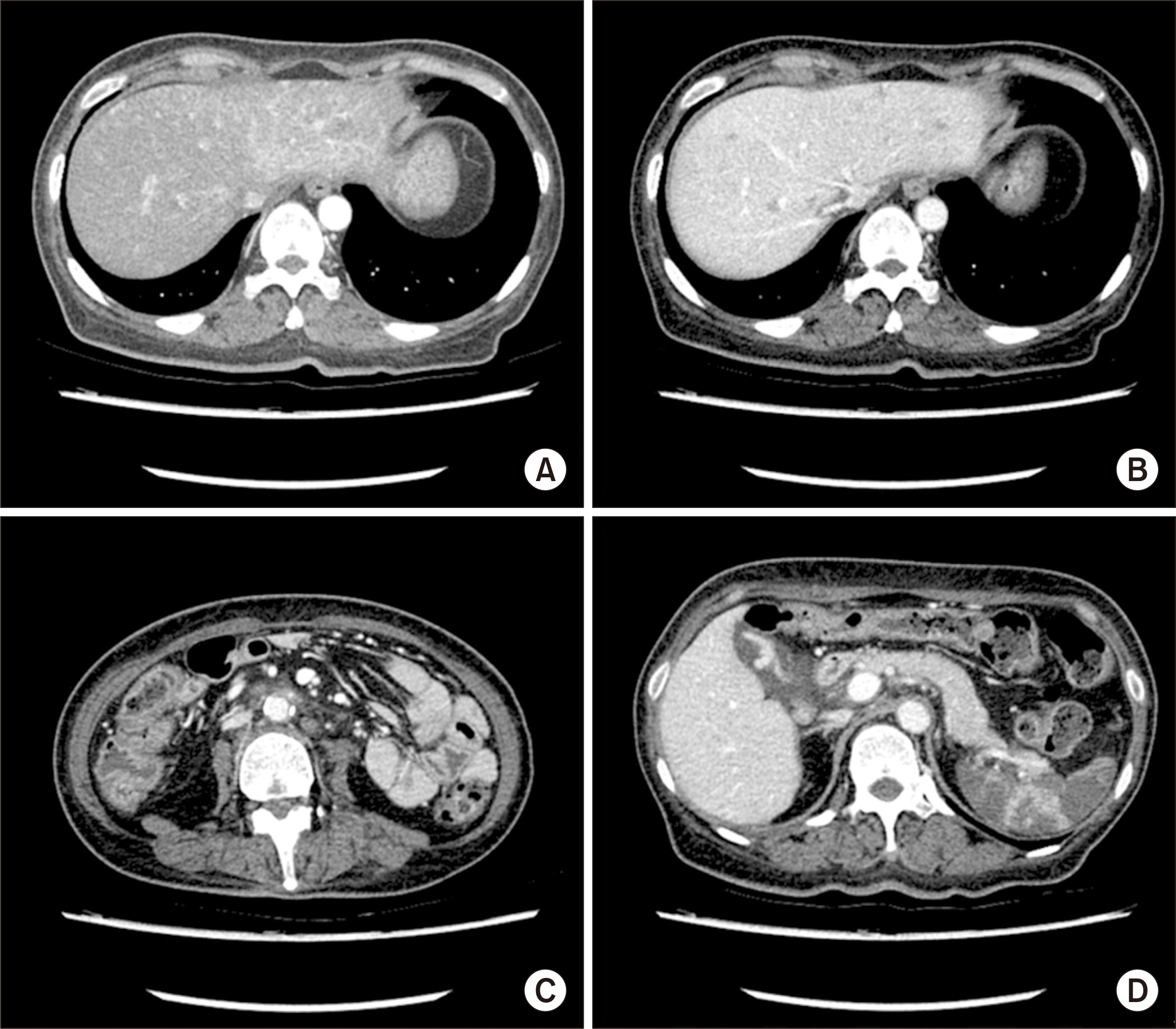
Eleven months following KT, the patient presented to the emergency department with symptoms of jaundice and abdominal pain. Physical examination revealed icteric sclerae and epigastric tenderness. Laboratory results were consistent with acute liver failure, including levels of aspartate transaminase (AST; 8,442 IU/L), ALT (5,972 IU/L), and total bilirubin (8.9 mg/dL) (Table 2). The patient’s EBV load was markedly elevated, at 6,467,257 copies/mL. Serologic tests for viral hepatitis and real-time polymerase chain reaction for cytomegalovirus returned negative results. APCT revealed multiple hepatic nodules, indicating PTLD with progressive liver involvement (Fig. 4). On the third day of hospitalization, the patient’s body temperature spiked to 39.8 °C. Empirical antibiotic treatment with ceftriaxone at a dosage of 2 g daily and metronidazole at 500 mg three times daily was initiated following blood and urine cultures, although no microorganisms were detected. A rescue chemotherapy regimen (cyclophosphamide [500 mg/m2, day 1], etoposide [50 mg/m2, day 1], vincristine [1.0 mg, day 1], and dexamethasone [4 mg every 8 hours, days 1–7]) was initiated on the 6th day of admission. Unfortunately, the patient died on day 12 of hospitalization.
DISCUSSION
We present a rare and aggressive case of EBV-associated monomorphic T-PTLD with a functioning allograft, occurring within 1 year of KT and resulting in the death of the patient. This case is notable because most T-PTLD cases develop several years after KT and are typically negative for EBV [3]. Given the fatal course and the absence of effective treatments for this condition, further research is necessary to improve the management of this rare disease. Based on two extensive review articles (which analyzed 107 and 58 cases of T-PTLD after KT) and additional case reports, most instances of T-PTLD manifest between 3 to 26 years following KT. Remarkably, only one case of EBV-negative monomorphic peripheral T-PTLD was reported within the first year after KT [3,6]. At our institution, of the 39 PTLD cases diagnosed, only five (12.8%) were T-PTLD (Table 3). Excluding the present case, these T-PTLD cases occurred later, ranging from 104 to 352 months after KT.
According to the World Health Organization’s 2016 classification, PTLD can be classified into two categories: polymorphic and monomorphic, with the latter including B and T/natural killer-cell subtypes [7,8]. Monomorphic PTLD is characterized by the monoclonal proliferation of malignant lymphocytes, while polymorphic PTLD features a mixture of B and T cells exhibiting destructive tissue infiltration [9,10]. Monomorphic T-PTLD tends to have a later onset and usually lacks the EBV genome within tumor cells [11]. However, in our case, the interval between transplantation and the development of T-PTLD was considerably shorter than what is commonly seen in T-PTLD cases. This discrepancy may be explained by the association of early-onset PTLD with EBV positivity, as well as extranodal involvement [12], both of which were observed in our case; nevertheless, further investigation is necessary to elucidate the mechanistic underpinnings of the relationship between disease onset and EBV serostatus. In the context of EBV positivity in this case, it has been noted that T cells expressing CD21 can be infected by EBV [13], and additional, as yet unidentified, receptors may facilitate EBV infection of T cells [14]. A prolonged and severe EBV infection could trigger an excessive immune response from T cells, and diminished immune surveillance by antitumoral T cells, due to immunosuppressive therapy, may contribute to the development of malignant T-PTLD [15]. Therefore, reduction of immunosuppression is the first-line treatment for PTLD. If this approach is not effective, second-line therapeutic options, including chemotherapy, radiotherapy, and surgical treatment, are considered [10]. In cases of B-PTLD with CD20 expression, rituximab may be an effective and safe option. However, if EB viremia worsens even after rituximab therapy, T-PTLD should be suspected, as in this case. Furthermore, preliminary studies have indicated promising clinical responses to adoptive immunotherapies, including EBV-specific cytotoxic T lymphocytes or donor lymphocyte infusion, although these therapies are not yet standard for PTLD treatment following solid organ transplantation [4]. Therefore, adoptive immunotherapies warrant consideration for treating EBV-associated PTLD, and additional studies are required to assess the efficacy of these therapeutic approaches.
Other immune-mediated or infectious diseases, such as hemophagocytic syndrome or EBV-associated fulminant hepatitis, were ruled out due to pathological findings indicative of PTLD. However, it is important to recognize that these conditions may be precipitated by EBV infection or a state of immunosuppression [16]. Given that our patient exhibited a range of nonspecific signs and symptoms, including fever, splenomegaly, cytopenia, and hypertriglyceridemia, these diseases should be taken into account in the differential diagnosis.
In conclusion, the prognosis of systemic T-PTLD remains poor due to its rarity and aggressive course. At present, no treatments are available for cases of T-PTLD that are refractory to conventional therapy. Consequently, additional research into the prevention and management of T-PTLD is warranted.
 XML Download
XML Download