

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Xenotransplantation refers to the transplantation of organs, tissues, cells, and the like across different species. The critical shortage of donor organs has led to the consideration of pig organs for this purpose. Pigs are deemed the most suitable candidates for xenotransplantation because they physiologically resemble humans, give birth to multiple offspring at once, and are easy to raise. However, substantial differences exist in molecular expression patterns between species, particularly in cell surface glycans, which are the carbohydrate components of glycoconjugates such as glycoproteins, glycolipids, or proteoglycans. These molecular discrepancies can elicit much stronger immune responses during xenotransplantation compared to allotransplantation. Galactose α1,3-galactose (α-Gal)- and N-glycolylneuraminic acid (Neu5Gc)-containing glycoconjugates exemplify glycans present in porcine vascular endothelial tissues but not in human tissues [1]. In early studies, transplanted organs such as hearts and kidneys were subjected to hyperacute rejection within minutes or hours. This was due to the presence of α-Gal, a sugar structure found on the surfaces of pig but not human cells. Natural antibodies in human blood that target this antigen bind to α-Gal on the endothelial cells of pig blood vessels. This binding activates the endothelial cells and triggers the complement system, leading to vascular damage and thrombus formation and constituting a hyperacute rejection response [2]. Consequently, the disparity in sugar structures between species is a primary factor inducing attacks by natural antibodies or innate immune cells. At present, research has pinpointed α-Gal and Neu5Gc as the key sugar structures on pig cell surfaces that bind to natural antibodies. Genetically engineered pigs have been created to eliminate these structures and to incorporate human molecules (CD46, CD55, or CD59) that inhibit complement activation. This genetic engineering has made it possible to overcome hyperacute rejection responses [3]. However, numerous sugar structures in these engineered pigs still differ from those in humans and can stimulate innate immune cells [4-6]. Additionally, pre-existing sugar structures may become more highly expressed, or new glycans may emerge, in genetically engineered pigs [4,6,7]. As a result, even with the use of current genetically engineered pigs, xenotransplantation may still face more intense inflammatory and innate immune responses than those observed in allotransplantation. The sugar structures can cause sustained systemic inflammation by continuously stimulating innate immune cells, including macrophages. In allotransplantation, inflammatory responses due to ischemia-reperfusion injury (IRI) during transplant preparation or surgical damage to the organ can be managed with anti-inflammatory agents, such as steroids. Due to the adverse effects of long-term steroid use, the drug dosage should be gradually decreased while monitoring the posttransplant prognosis. However, the sustained systemic inflammatory responses observed in xenotransplantation may preclude the gradual reduction of steroids [8]. Persistent systemic inflammation can further impair the function of the transplanted organ by interacting with the coagulation system. Therefore, it is uncertain whether immunosuppression protocols effective in allogeneic organ transplantation will also yield success in xenotransplantation.
Macrophages, as integral components of the innate immune system, play an important role in host defense. They achieve this by recognizing and responding to invading pathogens or their products during the initial stages of infection. This response includes activation, phagocytosis, induction of inflammatory responses, and antigen presentation to T cells. Although macrophages are crucial in the early inflammatory response to organ transplantation, they also contribute to the induction of transplantation tolerance [9]. Long-term transplant recipients who exhibit unresponsiveness to the transplanted organ often display graft-infiltrating macrophages with immunosuppressive properties [10]. A notable feature of macrophages is their plasticity, which enables them to easily switch between phenotypes in response to various physical and chemical stimuli from their environment [11]. Two well-established subpopulations of macrophages, each with distinct characteristics and functions, are the classically activated or inflammatory (M1) macrophages and the alternatively activated or anti-inflammatory (M2) macrophages. Recently, interest has been growing in regulatory macrophages (Mregs), a specialized subset that shares features with M2 cells and exhibits immunosuppressive capabilities. Human Mregs represent a unique state of macrophages capable of suppressing both inflammatory and T cell-mediated immune responses. The Hutchinson group in Germany has investigated the phenotype and immunological properties of Mregs derived from human peripheral blood mononuclear cells. Their findings highlight the clinical potential of these cells, demonstrating that pretransplant administration in allogeneic kidney transplantation can attenuate inflammatory and T cell-mediated immune responses, thereby reducing the need for prolonged and high-dose anti-inflammatory treatments [12]. Consequently, this therapeutic approach is anticipated to be even more effective in xenotransplantation, which involves substantially stronger immune responses. In this research, our objective was to explore this key obstacle currently facing xenotransplantation and to evaluate the utility of Mregs as a novel cell therapy to address the issue. We aimed to elucidate the principles and methods of the induction of human Mregs, define their typical phenotype, and examine their immunomodulatory functions. Furthermore, we discuss the mechanisms by which they exert their effects and the potential limitations on the practical application of this technology.
COAGULATION AND INFLAMMATION IN XENOTRANSPLANTATION
Recent evidence suggests that dysregulation of coagulation and persistent systemic inflammation are critical obstacles to successful pig-to-primate solid organ xenotransplantation. In the context of xenotransplantation, the primate coagulation system may be triggered by antibodies that target glycan antigens present on pig vascular endothelial cells, but not on primate cells [13-15]. Additionally, tissue factors (TFs) from both pig and primate sources contribute to the activation of coagulation [16,17], leading to the development of thrombotic microangiopathy [18,19]. Furthermore, incompatibilities between pig and primate molecules involved in the coagulation-anticoagulation systems exacerbate this dysregulation. Although pig thrombomodulin can bind to human thrombin, it does not effectively activate human protein C, an essential anticoagulant [20,21]. The pig TF pathway inhibitor is also insufficient in suppressing primate factor Xa, resulting in ineffective coagulation inhibition [22]. Pig von Willebrand factor spontaneously binds to primate platelet glycoprotein 1b and aggregates primate platelets, even without shear stress [23]. These activated platelets develop thrombosis after recruitment to damaged endothelial sites, leading to widespread activation of the coagulation system [24]. CD39 is an ectoenzyme present on the vascular endothelium that plays a vital role in preventing platelet aggregation triggered by extracellular adenosine triphosphate (ATP) and adenosine diphosphate. It accomplishes this by hydrolyzing these molecules into adenosine monophosphate and adenosine [25]. After transplantation, the expression of pig CD39 on vascular endothelial cells is reduced [26]. CD73, another enzyme involved in the metabolic process that converts ATP to adenosine, is expressed at much lower levels in pig endothelial cells compared to human cells, leading to decreased adenosine production and potentially causing intravascular coagulation [27]. Consequently, the introduction of human CD39 and CD73 genes into the pig genome has been suggested as a means to increase adenosine production. The expression of human CD39 in pigs has been demonstrated to significantly protect against myocardial damage in an ischemia-reperfusion model [28].
Inflammation is an unavoidable complication in solid organ transplantation, stemming from surgical trauma and IRI that occurs during the surgical procedure. Damage-associated molecular patterns (DAMPs) are released from injured tissues during IRI and are recognized by receptors on innate immune cells, which initiate inflammatory responses that can lead to graft rejection [29,30]. Therefore, early-phase suppression of inflammation in organ transplantation is critical for success. Despite this, recent research has shown that systemic inflammation can persist for several months following pig-to-primate solid organ xenotransplantation, even with the use of immunosuppressive therapy [8,31,32]. In recipients of heart and kidney transplants, serum levels of inflammatory cytokines and chemokines, such as tumor necrosis factor alpha (TNF-α), interferon gamma (IFN-γ), interleukin (IL)-12, and IL-18, are effectively managed by immunosuppressive therapy post-transplantation. However, serum levels of IL-6, monocyte chemoattractant protein 1 (MCP-1) [31], and C-reactive protein (CRP) [31-34] remain significantly elevated for an extended period. Notably, levels of these inflammatory mediators in nonhuman primate organ allograft recipients are significantly lower than those observed in organ xenograft recipients, suggesting that inflammatory responses are much more pronounced in xenograft recipients [31]. In the context of small artery patch xenotransplantation, recipient serum levels of inflammatory mediators also increased, apart from IL-6 and CRP, when no immunosuppressants were administered. When immunosuppressive medications were used, serum levels of inflammatory cytokines and chemokines were reduced, but levels of IL-6 and CRP remained high [31]. It has been hypothesized that IL-6, MCP-1, and CRP are involved in a positive feedback loop [35]. Given that IL-6 plays a pivotal role in chronic inflammatory diseases, autoimmune disorders, and cytokine storms [36-38], it likely occupies a central position in the sustained systemic inflammation seen in solid organ xenotransplantation. This inflammatory milieu contributes to the activation of monocytes, which then express TF [39], leading to the activation of coagulation [40,41]. The resulting amplification circuit between inflammation and coagulation promotes the release of additional inflammatory mediators and procoagulant factors [42], along with protease-activated receptor-mediated signaling and the further release of inflammatory cytokines. Therefore, preventing monocyte/macrophage-mediated inflammation therapeutically is critical for managing coagulation dysregulation following solid organ xenotransplantation.
REGULATORY MACROPHAGES
Macrophage Subtypes and Regulatory Macrophages Functions
Macrophages are phagocytic cells of the innate immune system that are essential for host defense, employing a variety of immunological and physiological mechanisms such as phagocytosis, inflammatory responses, antigen presentation, and wound healing. A notable feature of macrophages is their plasticity, which allows them to transition between subtypes [11]. In response to various stimuli, resting macrophages can undergo classical or alternative polarization to adopt distinct phenotypes. Two primary, functionally distinct macrophage subtypes are the classically activated macrophages (inflammatory macrophages, M1) and the alternatively activated macrophages (anti-inflammatory macrophages, M2) [43-46]. M1 macrophages arise in response to pathogen-associated molecular patterns or DAMPs at sites of infection or injury [47,48]. M2 macrophages, in turn, are polarized by macrophage colony-stimulating factor (M-CSF), IL-4, IL-10, and IL-13 [49]. Typically, M2 cells possess anti-inflammatory and regulatory properties, producing cytokines such as IL-4, IL-10, IL-33, and transforming growth factor beta (TGF-β) [50,51]. They demonstrate heightened phagocytic activity, express increased levels of scavenging, mannose, and galactose receptors, and produce greater concentrations of ornithine and polyamines due to an active arginase pathway. Additionally, they secrete substantial amounts of IL-10 and exhibit elevated levels of the IL-1 decoy receptor and IL-1Ra [52]. Overall, M2 macrophages exert anti-inflammatory effects and play a vital role in the antiparasite immune response necessary for parasite clearance, and they are involved in tissue remodeling, vasculogenesis, and tumor progression [11,44,53]. M2 cells can be identified by their distinct expression of surface proteins and secretion of various effector molecules [51]. M2 macrophages can be further categorized into subgroups M2a, M2b, M2c, and M2d [54-56]. M2a macrophages, also known as wound-healing macrophages, are stimulated by IL-4 and IL-13. They express high levels of the mannose receptor (also known as CD206), the decoy IL-1 receptor, and CCL17. They also secrete profibrotic factors such as TGF-β, insulin-like growth factor, and fibronectin, which aid in tissue repair [44,57-69]. M2b macrophages, or Mregs, are induced by toll-like receptor ligands [57,60-66] or IL-1R ligands [57,62,67-69] in conjunction with immune complexes or by various stimuli, including TGF, IL-10, or glucocorticoids [70]. They secret large quantities of IL-10 and small amounts of IL-12 [52,71], regulating both immune and inflammatory reactions. M2c macrophages are induced by IL-10 [45] or TGF-β [62] along with glucocorticoids. They are characterized by a strong anti-inflammatory profile, secreting large amounts of IL-10 and TGF-β, and express high levels of Mer receptor tyrosine kinase (MerTK), resulting in their efficient phagocytosis of apoptotic cells [62,72]. They are important in resolving inflammation, remodeling the extracellular matrix, and repairing tissue [73-75]. M2d macrophages, also referred to as tumor-associated macrophages, are induced by A2 adenosine receptor agonists or IL-6 [62,76-78]. These cells secrete high levels of IL-10, TGF-β, and vascular endothelial growth factor, but low levels of IL-12, TNF-α, and IL-1β [76,77,79-81], contributing to tumor angiogenesis, growth, and metastasis [62]. However, some overlap exists in the functional phenotypes of M2 macrophages. The subtypes of macrophages and their respective functions have been extensively covered in other studies [62,71,82].
Mregs are a type of macrophage known for their anti-inflammatory and immunosuppressive capabilities, sharing some of these properties with various M2 macrophage subsets [83-87]. The anti-inflammatory functions of Mregs are largely due to their capacity to inhibit M1 macrophages [84-86,88,89]. Mregs secrete high levels of the immunosuppressive cytokine IL-10, which curtails T cell proliferation [44,90], and promotes a T helper 2 response [12,91]. Conversely, Mregs produce minimal amounts of IL-12, a cytokine crucial for driving the differentiation of naïve T cells into T helper 1 cells, which are characterized by their robust production of IFN-γ. In human studies, Mregs have been shown to suppress mitogen-induced T cell proliferation in vitro [12] and to reduce T cell secretion of IL-2 and IFN-γ [91]. Additionally, they facilitate the induction of regulatory T cells (Tregs) through the release of TGF-β. Based on these distinctive immunosuppressive qualities, in vitro-generated Mregs have garnered interest as a potential cellular therapy for various inflammatory conditions, including sepsis [92], inflammatory bowel disease [93], autoimmune disorders [94,95] and organ transplantation [12,91].
Regulatory Macrophages Induction and Markers
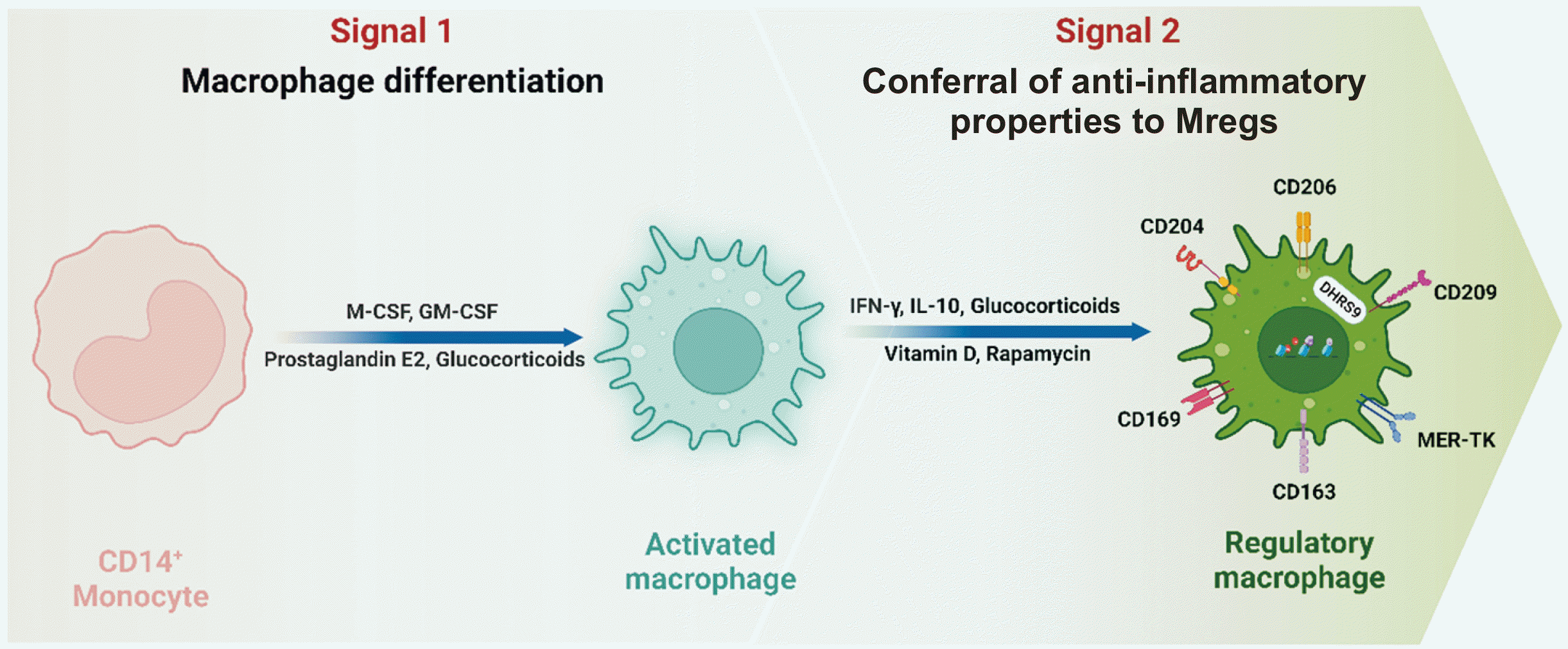
Mregs can be differentiated from monocytes and macrophages within a particular microenvironment [96]. Two signals from this microenvironment are required for Mreg induction. The first signal is elicited by exposure to M-CSF, granulocyte M-CSF, prostaglandin E2, glucocorticoids, or apoptotic cells [81,89]. The second can be achieved through stimulation with IFN-γ [97-99], IL-10 [100], or immunosuppressive reagents such as glucocorticoids [101], vitamin D [102,103], or rapamycin [104]. The initial signal facilitates the transformation of monocytes into macrophages, while the subsequent signal imparts immunosuppressive characteristics to these cells [62] (Fig. 1). Although various methods of Mreg induction have been examined, including our own approach (in which human monocytes were cultured on a three-dimensional micropatterned polydimethylsiloxane surface coated with polydopamine and the common cell adhesion peptide motif arginylglycylaspartic acid [RGD]) [105], as well as activation with a combination of phorbol 12-myristate 13-acetate, RGD, and vitamin D3 [103], the most extensively studied method involves combined treatment with M-CSF and IFN-γ. Human Mregs originate from CD14+ peripheral blood monocytes, which are first stimulated with M-CSF for 6 days and then with IFN-γ [99]. Typically, Mregs exhibit a fibrous morphology and express high levels of tissue-resident macrophage markers, such as CD163, CD169, CD204, CD206, CD209, and MerTK [106]. While identifying stable, specific markers for Mregs induced by various stimuli remains challenging, dehydrogenase/reductase 9 has been identified as a stable marker for human Mregs [106].
Mechanisms of Regulatory Macrophages Function and Limitations of Their Use
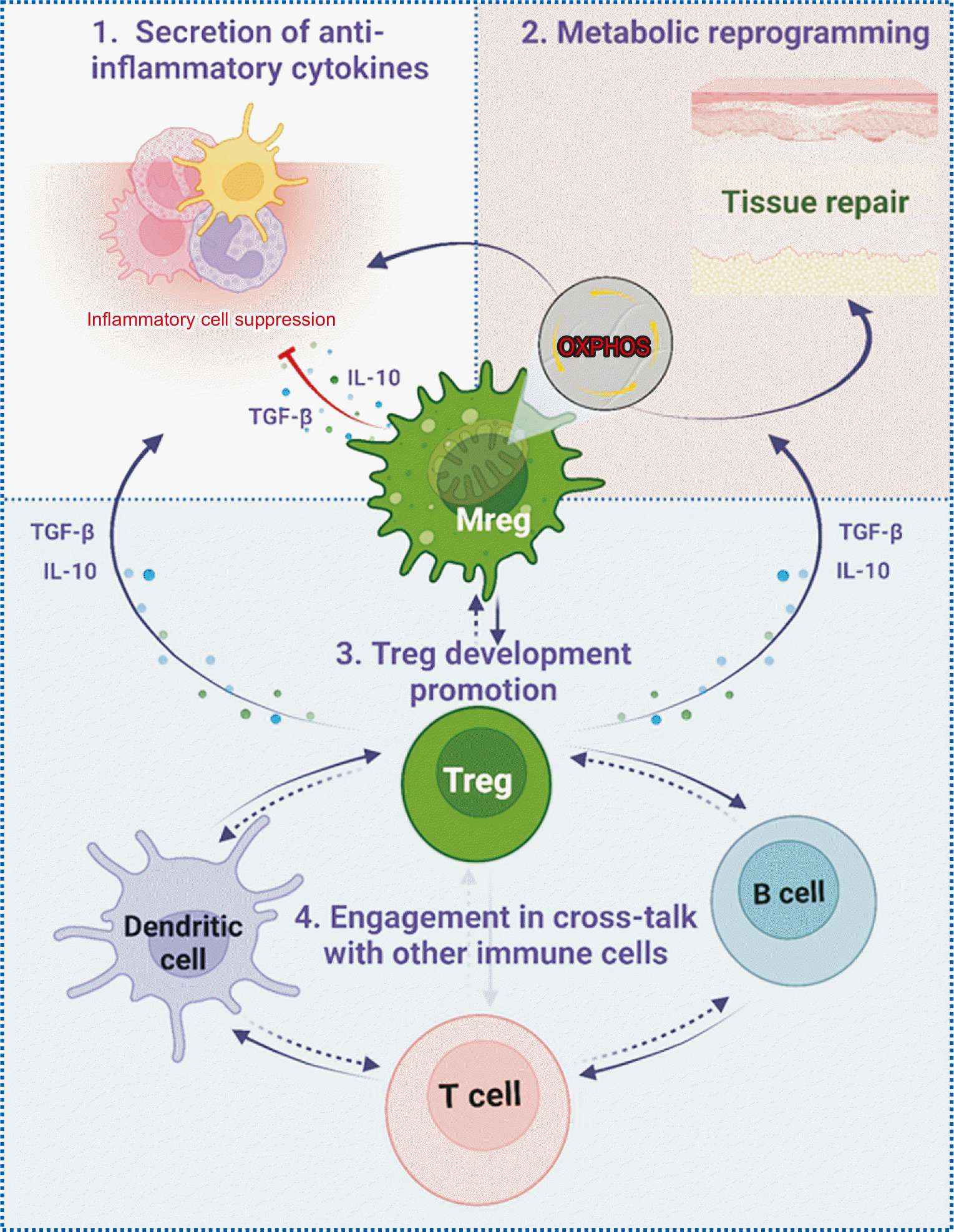
Although the mechanisms underlying the immunomodulatory functions of Mregs have not been fully elucidated, several have been identified: (1) Mregs secrete anti-inflammatory cytokines, such as IL-10 and TGF-β, which attenuate the proinflammatory response of other immune cells [91]; (2) Mregs can interact with and promote the development of Tregs, which possess potent suppressive capabilities to regulate immune responses and prevent immune-mediated tissue damage [52,91,103]; (3) Mregs undergo specific metabolic reprogramming, shifting their metabolism toward oxidative phosphorylation, a process associated with their anti-inflammatory and tissue-repair capabilities [107,108]; and (4) Mregs engage in cross-talk with other immune cells, such as dendritic cells, T cells, and B cells, thereby influencing their activation and function [52]. As such, Mregs contribute to a balanced and controlled immune response (Fig. 2).
Among these mechanisms, the interplay between Mregs and Tregs holds considerable importance in the modulation of the immune response at the site of graft transplantation. This modulation occurs through both direct and indirect interactions between these two cell types. Direct interactions involve cell-to-cell contact, in which T cells physically engage with Mregs, leading to the activation and proliferation of Tregs [12,91]. Indirectly, Mregs can influence Tregs by secreting anti-inflammatory cytokines, such as IL-10 and TGF-β. These cytokines are instrumental in enhancing the suppressive capabilities of Tregs, thereby promoting immune tolerance [91,109]. By interacting with one another and with other immune cells, Mregs and Tregs work in concert to preserve immune homeostasis.
One challenge in employing induced Mregs as a therapeutic approach for inflammatory diseases is their lack of stability. Numerous studies have shown that extended exposure to an inflammatory milieu can cause these cells to revert to a resting state or even transition to the proinflammatory M1 phenotype [110-114]. Furthermore, if the initial stimulants are not consistently provided, differentiated macrophages rapidly revert to their original phenotype [115]. Moreover, when a macrophage with one phenotype is exposed to a polarizing factor of the opposite type, it may tend to switch to a phenotype that aligns with the new activating environment [116]. Notably, the stability of these engineered cells is crucial to the success of any clinical therapy. Therefore, a deeper understanding of the mechanisms underlying macrophage plasticity is essential for developing therapeutic strategies that involve Mregs. The source of the precursor cells is another key factor in determining the fate of the activated cells. Selecting the appropriate cell source is vital for achieving a successful outcome in cell therapy. Research by various groups has indicated that macrophages with different origins exhibit unique responses to the same stimulant, leading to a range of phenotypes, each with specific functions [117]. Recent findings suggest that the propensity for phenotypic instability is largely due to the proliferation process of these macrophages [118,119]. The discovery of continuously maintained Mregs revealed that while newly formed cells lose their regulatory phenotype, the original parental macrophages retain their regulatory characteristics.
CONCLUSION
As a form of cellular therapy, the use of Tregs in organ allotransplantation offers considerable potential to improve transplant outcomes by promoting immune tolerance and reducing the likelihood of graft rejection. Nevertheless, xenotransplantation is hindered by the persistence of systemic inflammation, a critical contributor to immune rejection. Such inflammation may hinder the proliferation of Tregs and lead to increased cell death, thereby reducing the availability of these cells and undermining their capacity to suppress immune responses. Moreover, this inflammatory milieu may induce the conversion of Tregs into proinflammatory effector T cells, exacerbating the overall immune response and inflammation.
To address these challenges, it is imperative to explore alternative or complementary approaches. Mregs have emerged as a promising solution for mitigating systemic inflammation. Xenotransplantation presents certain advantages over allotransplantation, such as the potential for genetic modification and the opportunity for pre-conditioning prior to the transplant. Therefore, by gaining a thorough understanding of the precise mechanisms governing the generation, stability, and immunomodulatory functions of Mregs, the application of Mregs generated in vitro stands poised to reduce the dosage and duration of the anti-inflammatory and immunosuppressive drugs required in future preclinical and clinical settings.
 XML Download
XML Download