

 PDF
PDF Citation
Citation Print
Print


TO THE EDITOR: We found the comprehensive review article by Park et al. [1], which focused on T-cell large granular lymphocytic (T-LGL) leukemia and its typical low-grade bone marrow involvement, to be very enlightening. T-LGL leukemia can potentially lead to serious complications, such as severe infections due to neutropenia or the occurrence of pure red cell aplasia (PRCA). Herein, we present a case report of T-LGL leukemia accompanied by PRCA. Additionally, we include focused reviews on leukemic T/natural killer (NK)-cell lymphomas with frequent bone marrow infiltration and on acquired PRCA associated with different etiologies.
CASE
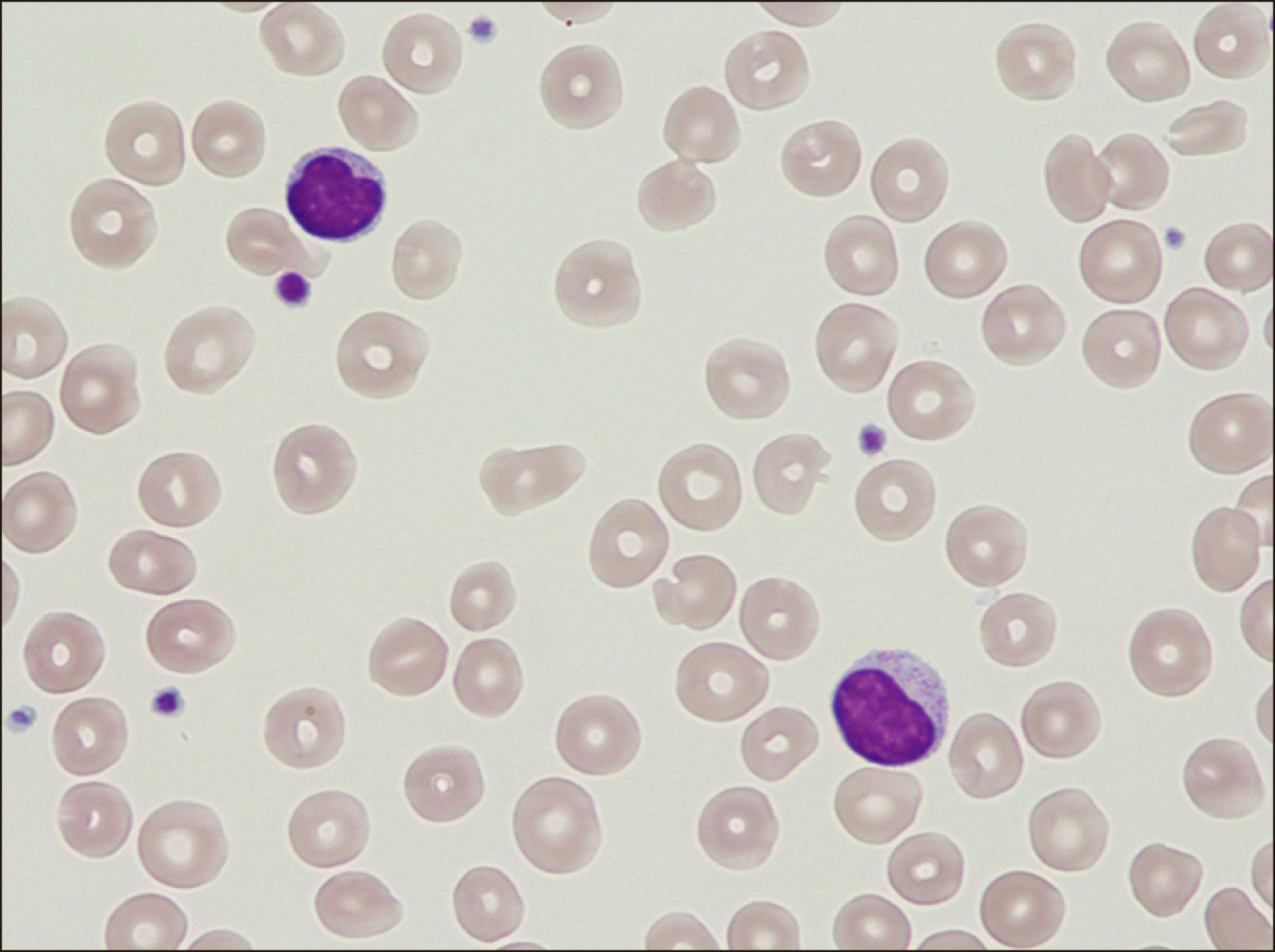
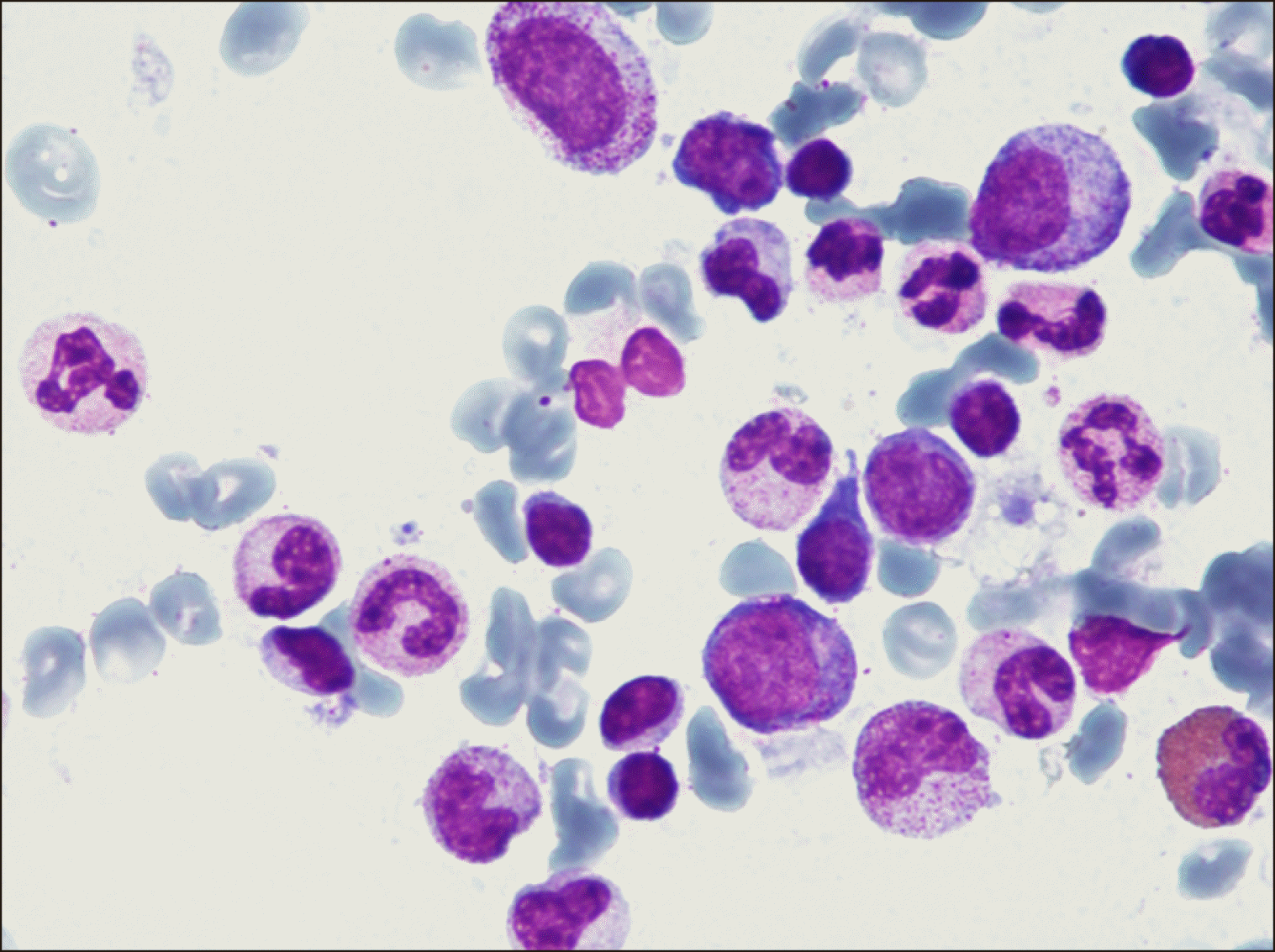
An 85-year-old male with unexplained transfusion-dependent macrocytic anemia prompted us to perform a bone marrow biopsy. Cytological and cytogenetic analyses did not confirm the primary differential diagnosis of suspected myelodysplastic syndrome (MDS), and next-generation sequencing of 30 genes associated with myeloid neoplasms yielded negative results. By contrast, erythroblastopenia together with the presence of atypical lymphocytes with azurophilic cytoplasmic granules was observed (Fig. 1). Immuno-cytological assessment revealed an aberrant T-cell profile (CD2+/CD3+/ CD8++/CD57+/CD5dim/CD7-). Moreover, determination of the clonal T-cell status through T-cell receptor rearrangement analysis led to the diagnosis of T-LGL [1]. Additionally, peripheral differential blood examination revealed reticulocytopenia (regarded as a hallmark of acquired adult PRCA) in addition to erythropoietic precursor paucity (Fig. 2) [2]. Although the degree of bone marrow infiltration in our patient was low (12% lymphoma cells), concomitant PRCA was evident. Owing to his age and comorbidities, he was treated with red blood cell transfusions and corticosteroids. The patient visits the outpatient clinic weekly or biweekly, and no infections and other major complications have been reported.
DIFFERENTIAL DIAGNOSIS OF LEUKEMIC T/NK-CELL LYMPHOMAS
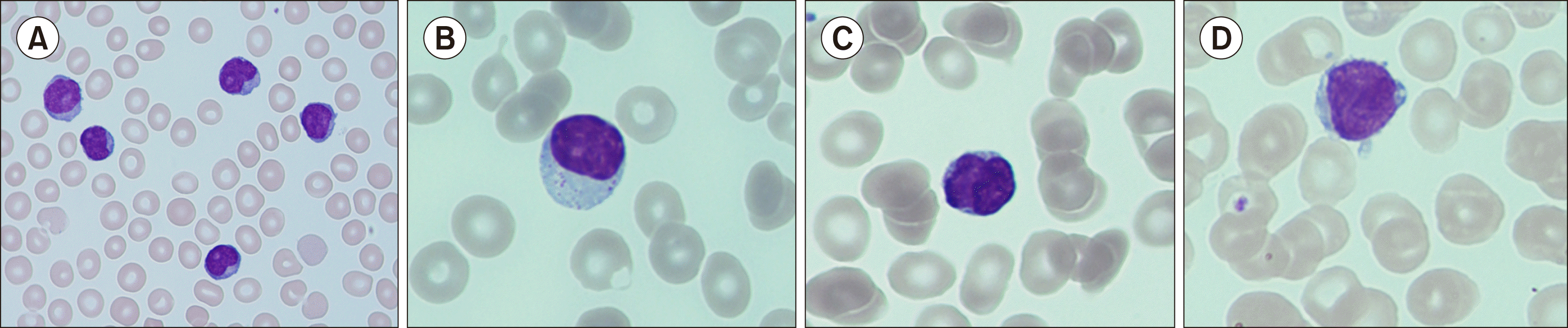
The World Health Organization classification of hematolymphoid tumors includes many types of T/NK-cell lymphoma. However, T/NK-cell lymphomas with a characteristic leukemic course and bone marrow involvement, respectively, are predominantly restricted to T-LGL leukemia, T-prolymphocytic leukemia (T-PLL), natural killer cell-large granular lymphocytic (NK-LGL) leukemia, adult T-cell lymphoma (ATCL), Sezary syndrome (SS), and aggressive natural killer cell leukemia (ANKL) [3]. For differentiating the T/NK-cell lymphomas, immunocytological analysis in combination with cytomorphological investigations remains pivotal. Cytogenetic and molecular genetic investigations are unable to show recurrent mutations or aberrations with proper disease specificity and are more useful for gaining prognostic information. However, the assessment of T-cell clonality as a precondition for the reliable diagnosis of T-cell lymphomas is still achieved through genetic analyses. Skin lesions, exanthema, and lymphadenopathies are frequently observed in the above-mentioned lymphomas, with erythroderma occurring the most in SS. Among the possible conditions that can occur with mature T/NK-cell lymphomas, lymphoproliferative disorders (leukocytosis or lymphocytosis) are typically seen in T-PLL, ATCL, and SS, whereas cytopenias are more prevalent in T-LGL leukemia, NK-LGL leukemia, and ANKL. Hyperleukocytosis and an aggressive lymphocyte doubling time are characteristics of T-PLL, which was formerly named T-chronic lymphatic leukemia [4-7]. Morphological analysis remains a valuable diagnostic tool for differentiating and classifying these lymphomas, with microscopic investigation of blood smears being an important screening procedure. Although the morphological appearance of the T-cell lymphomas varies widely, certain features are frequently associated with distinct disease types. The nuclei often show convolutions and multilobulation in an impressive cloverleaf form in ATCL; the cells are usually darker and appear cerebriform in SS; prominent nucleoli are pathognomonic in T-PLL; and cytoplasmic granules can be detected in T-LGL leukemia, NK-LGL leukemia, and ANKL (Fig. 3).
In ANKL, neoplastic cells can present with finely dispersed chromatin, mimicking a blast-like morphology as an expression of the higher malignant state of the disease. Immunocytological investigations have shown that the CD4/CD8 profile can guide the preliminary classification of mature T-cell lymphomas. The CD4/CD8 ratio is always substantially increased in T-PLL, ATCL, and SS, whereas the CD8 expression level is negative or markedly decreased (with the CD4/CD8 ratio consequently reduced) in T-LGL leukemia. CD16, CD56, and CD57 show variable but usually positive expression levels in the different types of LGL lymphomas. T-LGL leukemia usually shows positivity for the pan-T-cell marker, whereas NK-LGL leukemia and ANKL are negative for CD3. The CD30-positive immuno-phenotype of ANKL is indicative of the aggressive behavior of this lymphoma. Thus, the diagnosis of leukemic T-cell lymphomas relies strongly on cytomorphological and immunocytological investigations and the exclusion of common mature T/NK-cell lymphomas. An overview of the diagnostic characteristics of the T/NK-cell lymphomas is presented in Table 1 [4-7].
ACQUIRED PURE RED CELL APLASIA
With regard to the hematological malignancies, lymphoproliferative disorders are commonly considered the causes of PRCA. According to Vlachaki et al. [8], T-cell lymphomas (T-LGL leukemia and angioimmunoblastic T-cell lymphoma) appear to be a more frequent cause of PRCA than B-cell lymphomas. The timing of PRCA manifestation in patients with lymphoma is difficult to predict, as it may precede the lymphoma, present simultaneously with it, or persist after disease remission [8]. For individuals with concomitant PRCA and lymphoma, the recommended treatment is an immunosuppressive and/or chemotherapeutic therapy regimen that can include corticosteroids, cyclophosphamide, and cyclosporin. In patients with therapy refractoriness, the monoclonal anti-CD20 antibody rituximab and the monoclonal anti-CD52 antibody alemtuzumab can induce remission [8]. Methotrexate is regarded as the first-line treatment for patients with T-LGL leading to neutropenia, whereas cyclophosphamide and cyclosporin are preferred for those with concomitant T-LGL and PRCA [1]. Generally, STAT3 mutations are prevalent in PRCA, with a mutation frequency of over 40% in PRCA associated with T-LGL leukemia. However, STAT3 mutations are not exclusive to PRCA-associated lymphomas, as they have also been reported in other hematological malignancies such as MDS and aplastic anemia. Regardless of the underlying PRCA-associated neoplasm, patients with STAT3 mutations characteristically develop impaired responsiveness to cyclosporin and a higher likelihood of therapy resistance compared with those carrying wild-type STAT3 [9, 10].
Because PRCA is a possible prodromal phase of MDS, Fujishima et al. [9] conducted a sequencing analysis of myeloid genes and proposed KDM6A, TET2, and DNMT3A mutations as putative drivers of PRCA. They concluded that clonal hematopoiesis by progenitor and stem cells might be related to the pathophysiology of chronic PRCA in at least one subgroup of patients. However, most of these variants were found in epigenetics-related genes, reflecting possible age-related hematopoietic alterations. However, further clinical trials are required to confirm these findings [9]. In MDS, abnormal stem cell differentiation and immune alterations are the main causes of PRCA development, and this pathophysiology determines the treatment approach. In a case series comprising six patients with concomitant MDS and PRCA, each patient received a different treatment regimen, but all of them were administered erythropoietin [11]. Complete remission was achieved in two patients, one of whom had additionally received corticosteroids, cyclosporin A, and intravenous immunoglobulin while the other had received androgen and thalidomide [11].
In 2016, Korde et al. [12] conducted the first systematic investigation of the relationship between PRCA and plasma cell dyscrasias. They reported a 24% prevalence of monoclonal gammopathies among 51 patients diagnosed with PRCA and added a paraprotein-related phenomenon to the spectrum of possible PRCA mechanisms. Interestingly, all patients were diagnosed with pre-stage myelomas (e.g., smoldering myeloma or monoclonal gammopathy of undetermined significance) and lacked end-organ damage (bone lesions) or myeloma-associated complications. Paradoxically, the bone marrow exhibited hypercellularity, eosinophilia, and increased reticulin fibrosis. Although anemia is a key feature of myeloma, whether or not modulation of the tumor microenvironment by paraproteins is the cause of PRCA remains speculative. Anemia in patients with myeloma is usually accompanied by regular maturation and is a result of iron dysregulation, renal insufficiency, or marrow replacement in cases of higher infiltration of atypical plasma cells. This is in contrast to the common concept of predominantly T-cell-driven PRCA pathomechanisms [12]. The study by Korde et al. [12] also provided data on the treatment of eight patients with monoclonal gammopathy and PRCA. All patients were refractory to immunosuppressive therapies such as cyclosporin A, rituximab, and corticosteroids. Six patients showed no response to daclizumab, an antibody for idiopathic PRCA. In three patients, anti-myeloma therapy (lenalidomide and bortezomib) led to remission with transfusion independence [12].
In conclusion, the diagnosis of T-LGL leukemia is based on immunocytological and cytomorphological investigations. Subsequent molecular genetic analyses are important for confirming the diagnosis of T-cell neoplasias based on T-cell receptor clonality. PRCA is a serious complication of T-LGL leukemia.
 XML Download
XML Download