

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
The ‘endothelium’ is a thin layer of vascular endothelial cells that lines blood vessels and plays a central role in regulating vascular tone, blood fluidity, and flow [1]. Endothelial dysfunction involves abnormal shear stress and a pro-inflammatory response, which can cause various cardiovascular diseases (CVDs) such as hypertension, atherosclerosis, and stroke [1]. Generally, chronic and excessive oxidative stress is considered a major cause of endothelial dysfunction. Stacked reactive oxygen species (ROS) can promote endothelial cell apoptosis, vascular permeability, and leukocyte adhesion to arterial walls [2]. In patients with coronary atherosclerosis, endothelial superoxide production and nicotinamide adenine dinucleotide phosphate oxidase expression have been clearly associated with lesion severity [3]. Reduced activity of superoxide dismutase 2, an antioxidant enzyme for superoxide removal, increased inflammatory cell infiltration into atherosclerotic plaques and decreased plaque stability during aging in apolipoprotein-E-deficient mice [4]. Therefore, reducing endothelial ROS production seems to be one of the best therapeutic strategies for preventing CVDs.
G protein-coupled receptor 40 (GPR40), which was first reported as a protein with seven transmembrane domains at human chromosomal locus 19q13.1 [5], is considered an orphan receptor for several fatty acids. Itoh et al. [6] reported that docosahexaenoic acid, linoleic acid, and oleic acid, which are long-chain fatty acids, can be natural ligands for GPR40. Therefore, GPR40 was named free fatty acid receptor 1 (FFAR1). Recently, GPR40 has received attention as a therapeutic target for type 2 diabetes mellitus. GPR40 deficiency increased blood glucose level by lowering insulin secretion in obese mice [7], whereas GPR40 overexpression improved glucose-stimulated insulin secretion, inhibiting hyperglycemia [8]. As a result, many GPR40-specific agonists have been developed as anti-hyperglycemic agents [9]. Several researchers have reported that GPR40 activation was associated with beneficial effects against metabolic stress. Treatment with CNX-011-67, a GPR40-specific agonist, not only increased insulin secretion, but also reduced the inflammatory response, oxidative stress, and apoptosis in pancreatic β-cells [10]. Treatment with a GPR40 agonist can induce the phosphorylation of adenosine monophosphate-activated protein kinase, which is a representative anti-inflammatory molecule [11], in hepG2 cells [12] and inhibit obesity-mediated hepatic lipid accumulation and collagen deposition [13]. In addition, lipopolysaccharide-induced adhesion molecules and monocyte attachment were blocked in vascular endothelial cells treated with LY2922470, a GPR40 agonist [14]. Based on those results, we hypothesized that endothelial oxidative stress could be regulated by GPR40 activation.
To discover whether GPR40 agonists had beneficial roles against palmitate-mediated oxidative stress in vascular endothelial cells, we examined whether treatment with GPR40 agonists (1) induces the expression of antioxidant molecules such as nuclear factor erythroid 2–related factor 2 (NRF2) and heme oxygenase-1 (HO-1); (2) reduces palmitate-mediated superoxide production; (3) inhibits the enhancement of endoplasmic reticulum (ER) stress-related molecules under palmitate treatment; and (4) reduces palmitate-induced nuclear damage and cell death.
METHODS
Cell culture and reagents
Human umbilical vein endothelial cells (HUVECs, Thermo Fisher Scientific, Waltham, MA, USA) were cultured in 0.2% gelatin-coated dishes with endothelial cell medium (ScienCell, Carlsbad, CA, USA) containing endothelial cell growth supplement (ScienCell) and 5% fetal bovine serum (ScienCell). The GPR40-partial agonists LY2922470 and TAK875 (MedChemExpress, Monmouth Junction, NJ, USA), GPR40-full agonist AM1638 (MedChemExpress), and the NRF2 inhibitor ML385 (Sigma Aldrich, St. Louis, MO, USA) were dissolved in dimethyl sulfoxide (Sigma Aldrich). Palmitate (Sigma Aldrich) was dissolved in ethanol to concentration of 400 mM, mixed with 10% fatty acid-free bovine serum albumin (BSA, Sigma Aldrich) solution to prepare 4 mM palmitate-BSA solution, and then incubated at 37°C for 1 hour. The 10% fatty acid-free BSA containing equal ethanol were used as a vehicle.
Western blotting and antibodies
Total proteins from HUVECs were extracted using PRO-PREP (iNtRON Biotechnology, Seongnam, Korea) and quantified using the Bradford assay. Equal amounts of proteins were electrophoresed on 8% to 15 % polyacrylamide gels and moved to nitrocellulose (NC) membranes (Amersham Bioscience, Waltham, MA, USA). The NC membranes were soaked sequentially with 5% nonfat dry milk or 5% BSA, primary antibody, and horseradish peroxidase–conjugated secondary antibody in Tris-buffered saline containing 0.05% Tween 20. The membranes were reacted with enhanced chemiluminescence substrate (Bio-Rad Laboratories, Hercules, CA, USA).
The antibody sources were anti-β actin mouse immunoglobulin G (IgG), anti-lamin B1 mouse IgG, anti-total inositol requiring enzyme 1α (IRE1α) rabbit IgG (Santa Cruz Biotechnology, Santa Cruz, CA, USA); anti-p-IRE1α rabbit IgG (Thermo Fisher Scientific); anti-NRF2 rabbit IgG, anti-Kelch-like ECH-associated protein 1 (Keap1) rabbit IgG, anti-HO-1 rabbit IgG, anti-total protein kinase R-like endoplasmic reticulum kinase (PERK) rabbit IgG, anti-p-PERK rabbit IgG, anti-CCAAT/enhancer‐binding protein homologous protein (CHOP) mouse IgG, anti-spliced X-box binding protein-1 (XBP-1s) rabbit IgG, anti-cleaved caspase 3 rabbit IgG, anti-poly(ADP-ribose) polymerase (PARP)-1 rabbit IgG (Cell Signaling Technology, Danvers, MA, USA); and anti-nicotinamide adenine dinucleotide phosphate: quinone oxidoreductase 1 (NQO1) mouse IgG, and anti-GPR40 goat IgG (Abcam, Cambridge, UK).
GPR40 knockdown
Scrambled and GPR40 small interfering RNAs (Santa Cruz Biotechnology) were inserted into separate samples of HUVECs using Lipofectamine reagent (Thermo Fisher Scientific) according to the user manual.
NRF2 fractionation
Proteins were separated into cytosolic and nuclear fractions using a protein fractionation kit (Cayman Chemical, Ann Arbor, MI, USA) according to the manufacturer’s directions. Each fraction was analyzed by Western blotting to confirm the localization of NRF2.
Superoxide staining
Intracellular superoxide was stained using dihydroethidium (DHE; red florescence dye, Thermo Fisher Scientific) according to the user manual. The red fluorescence level was observed under a fluorescence microscope (Olympus, Tokyo, Japan) and was quantified using ImageJ software (National Institutes of Health, Bethesda, MD, USA). Blue fluorescence indicates nuclei. The relative mean fluorescence intensity value of DHE was calculated by flow cytometry (Beckman Coulter, Krefeld, Germany).
Cell viabilities
HUVECs were stimulated with EZ-CYTOX solution (Daeil Lab Service, Seoul, Korea) for 1 hour, after which cell viability was measured using a 450 nm wavelength filter on a microplate reader (Bio-Rad Laboratories).
Nuclear morphology
HUVECs were fixed in 3.7% formaldehyde for 15 minutes and then stained with Hoechst dye (Thermo Fisher Scientific) for 15 minutes. Images of the nuclear morphology were obtained using a fluorescence microscope (Olympus), after which the ratio of damaged to healthy cells was calculated.
DNA fragmentation
A terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay kit (Mybiosource, San Diego, CA, USA) was used according to the user manual to confirm DNA fragmentation. Damaged cells labeled with red fluorescence were observed and counted under a fluorescence microscope (Olympus).
RESULTS
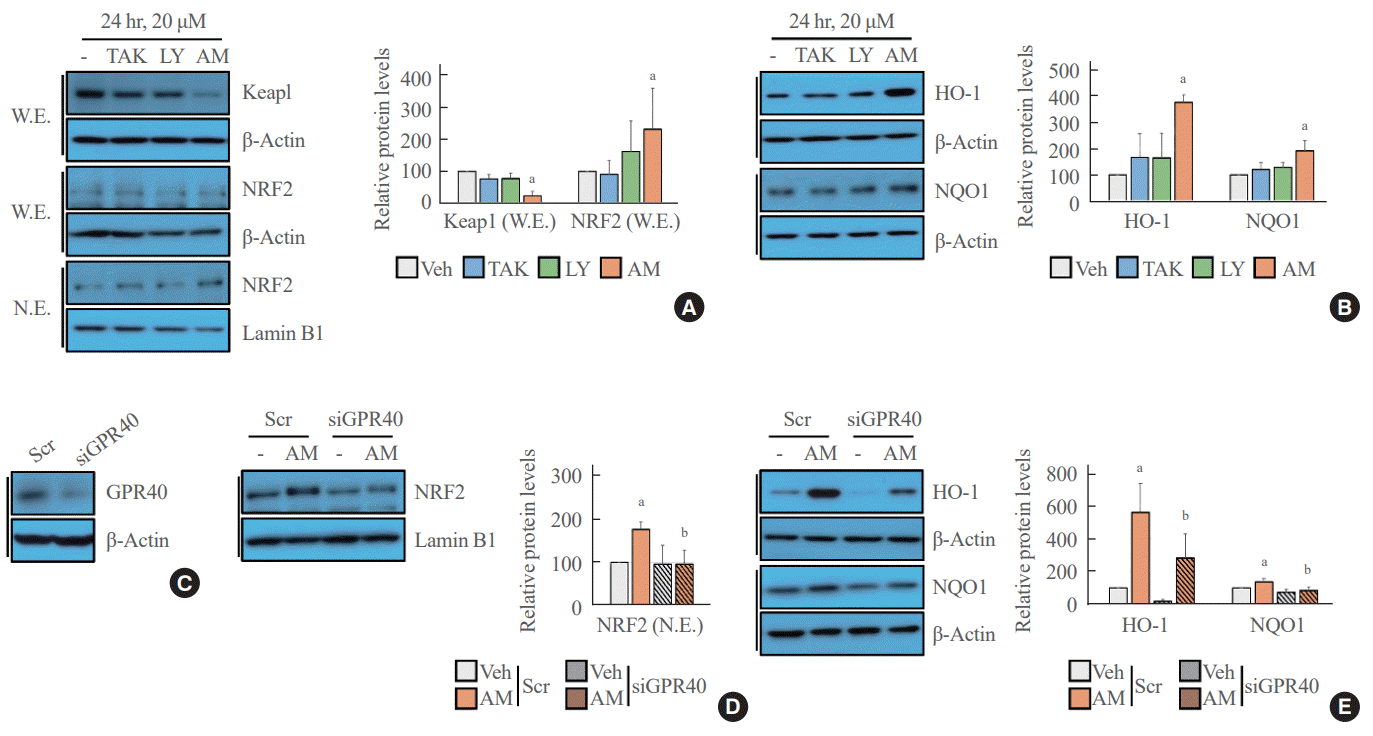
NRF2-mediated signaling was activated after treatment with AM1638
The Western blot analysis showed that treatment with AM1638, a full agonist of GPR40, significantly reduced Keap1 level and increased NRF2 translocation from the cytoplasm to the nucleus; however, treatment with LY2922470 or TAK875, partial agonists of GPR40, did not affect Keap1 level and NRF2 translocation to the nucleus (Fig. 1A). Similarly, the NRF2 target genes HO-1 and NQO1 increased after AM1638 treatment but not after LY2922470 or TAK875 treatment (Fig. 1B). To determine whether the effects of AM1638 are GPR40-dependent or GPR40-independent, we treated GPR40-knockdown HUVECs with AM1638. AM1638-mediated NRF2 translocation to the nucleus and HO-1 and NQO1 expression were canceled by GPR40 silencing (Fig. 1C-E).
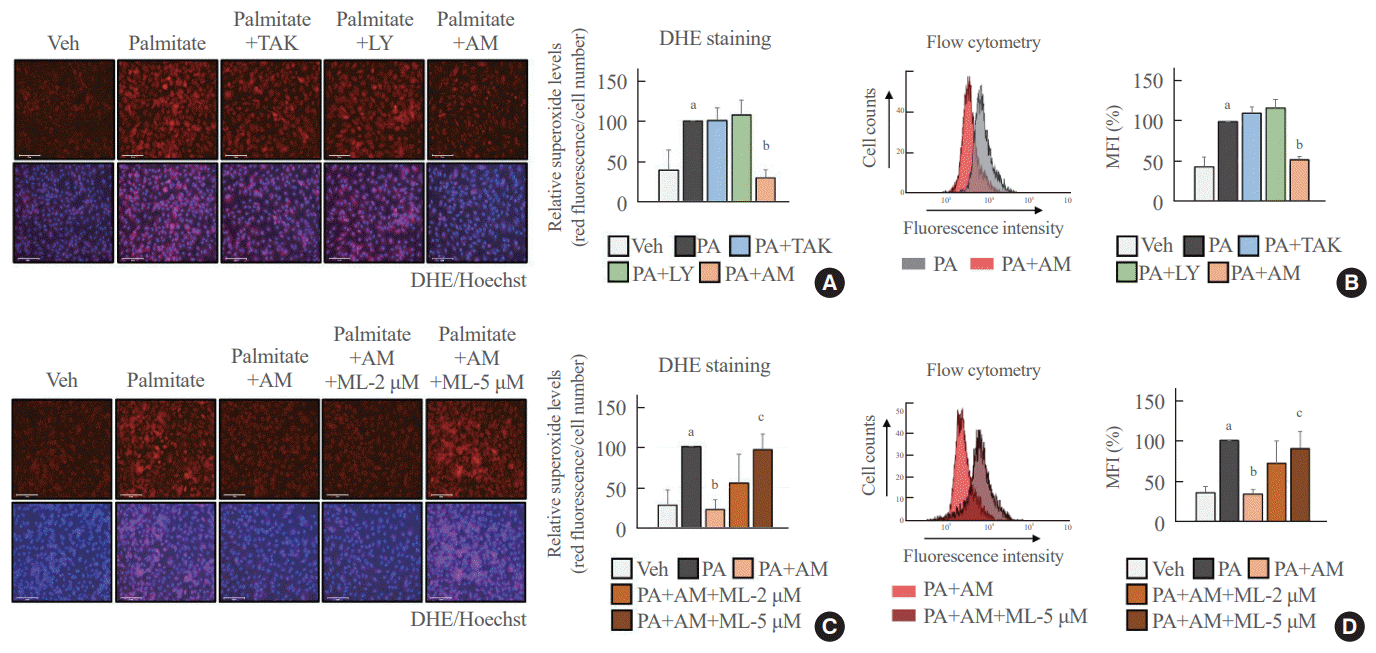
Palmitate-induced superoxide production was blocked in an NRF2-dependent manner after treatment with AM1638
We determined the antioxidant role of GPR40-specific agonists in palmitate-treated HUVECs. The fluorescence images and flow cytometry analysis showed that AM1638 meaningfully reduced palmitate-mediated superoxide production, whereas LY2922470 and TAK875 did not (Fig. 2A, B). Treatment with ML385, an NRF2 inhibitor, abolished the AM1638-mediated reduction in superoxide levels (Fig. 2C, D).
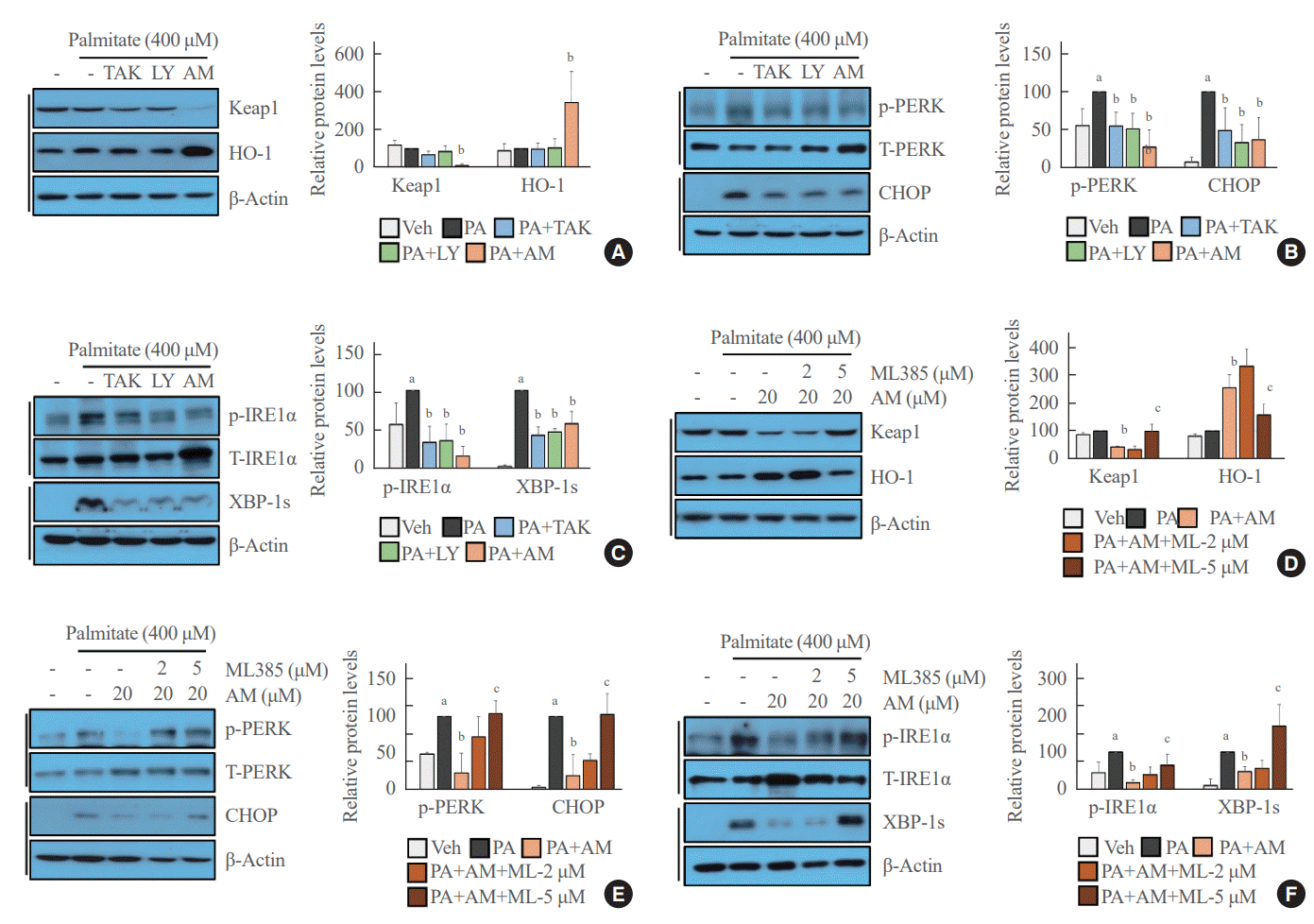
Palmitate-mediated ER stress was inhibited by GPR40 agonists in NRF2-dependent and -independent manners
We revealed that GPR40-specific agonists play a beneficial role against palmitate-mediated endothelial ER stress. Under palmitate treatment, AM1638 reduced Keap1 expression and induced HO-1 expression, whereas LY2922470 and TAK875 had no effect on Keap1 and HO-1 expression (Fig. 3A). Palmitate-induced PERK phosphorylation and CHOP expression were reduced not only after treatment with AM1638, but also after treatment with LY2922470 or TAK875 (Fig. 3B). Similarly, all three agonists inhibited IRE1α phosphorylation and XBP-1 splicing under palmitate treatment (Fig. 3C). The AM1638-mediated effects such as reduced Keap1 expression, increased HO-1 expression, decreased PERK/CHOP and IRE1α/XBP-1s pathways, were all canceled after treatment with ML385 (Fig. 3D-F).
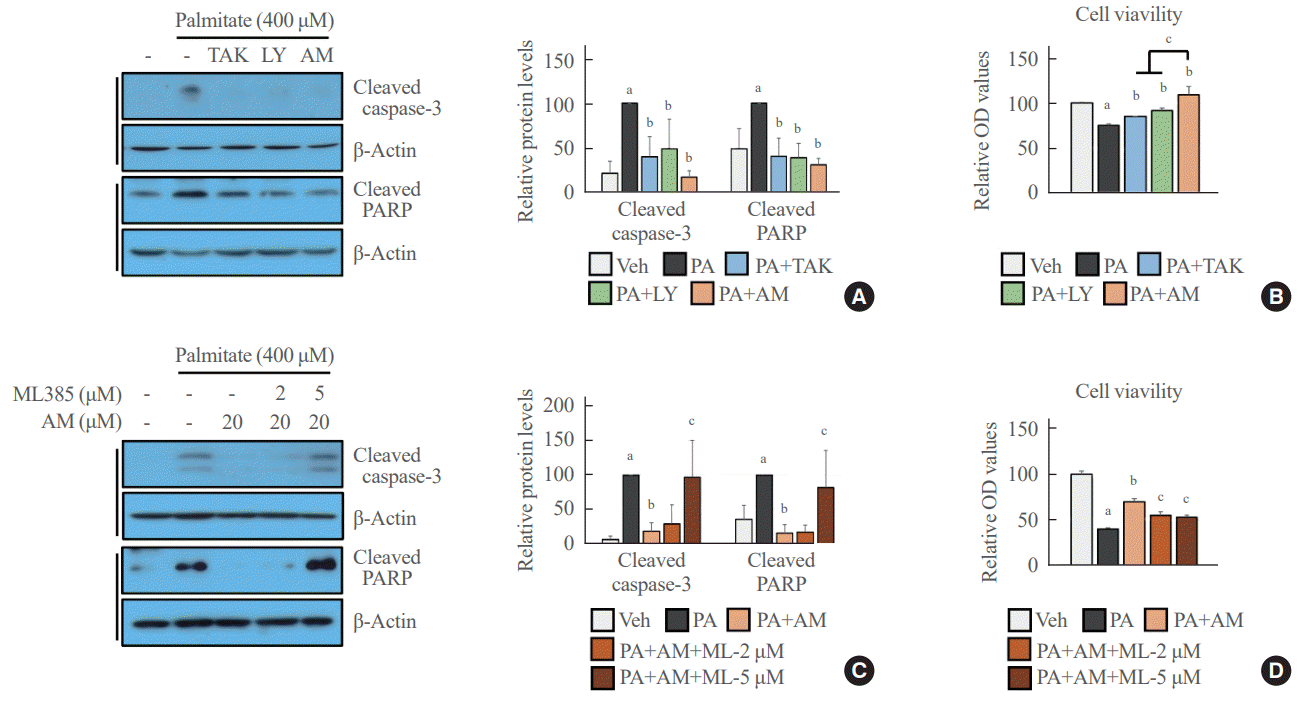
GPR40 agonists inhibited palmitate-mediated cytotoxicity in NRF2-dependent and independent manners
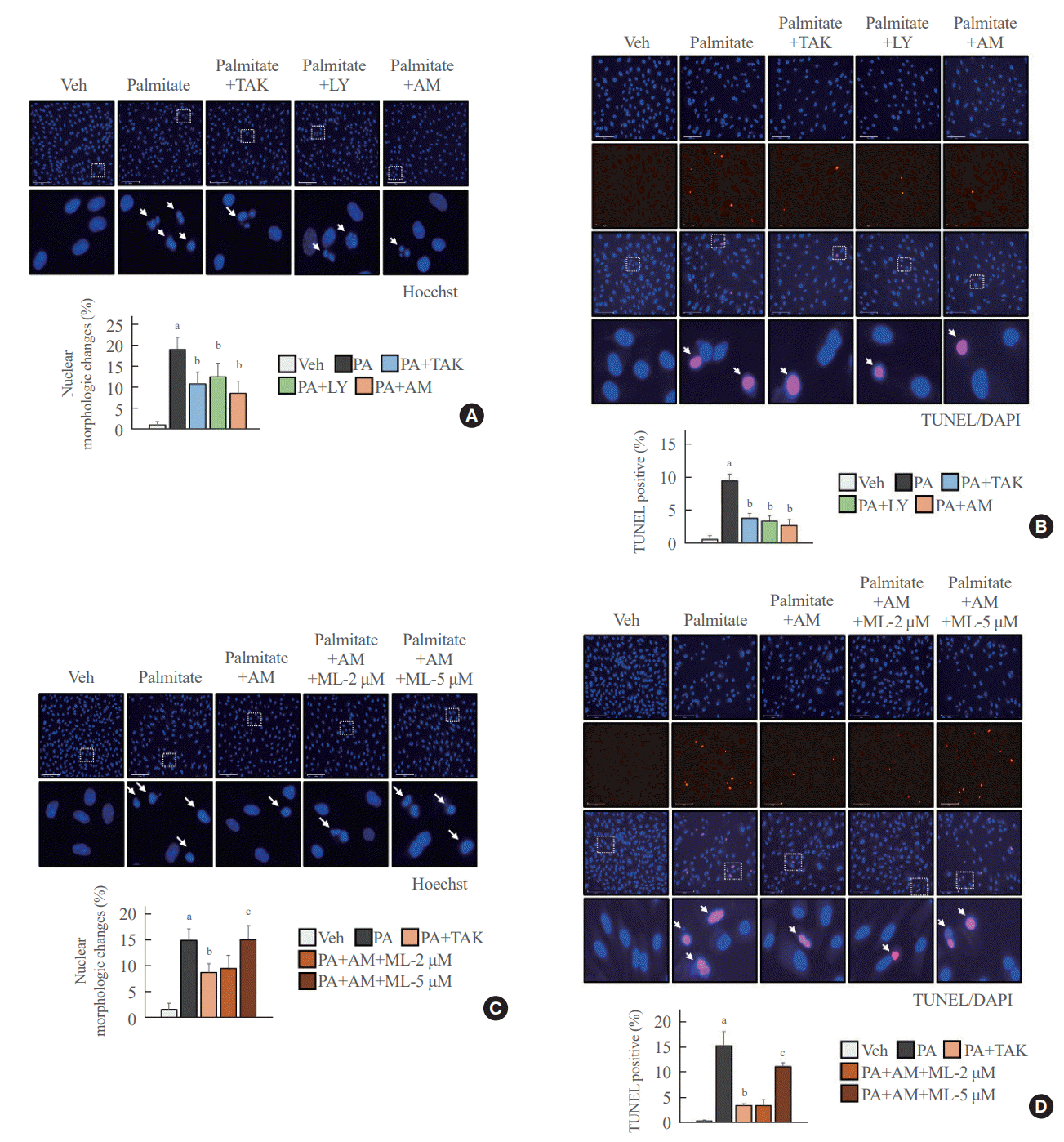
AM1638, LY2922470, and TAK875 all had protective effects against palmitate-mediated endothelial cytotoxicity. In HUVECs, all three agonists reduced palmitate-induced caspase 3 and PARP cleavage (Fig. 4A), restoring cell viability (Fig. 4B). AM1638 treatment increased cell viability more than treatment with either of the two partial agonists, irrespective of caspase 3 and PARP cleavage. The AM1638-mediated abolition of caspase-3/PARP signaling and improvement in cell viability were reversed by ML385 treatment (Fig. 4C, D). Furthermore, palmitate-induced nuclear condensation (Fig. 5A) and fragmentation (Fig. 5B) were significantly decreased in HUVECs stimulated with GPR40 agonists. Treatment with ML385 abolished the AM1638-mediated recovery of nuclear damage (Fig. 5C, D).
DISCUSSION
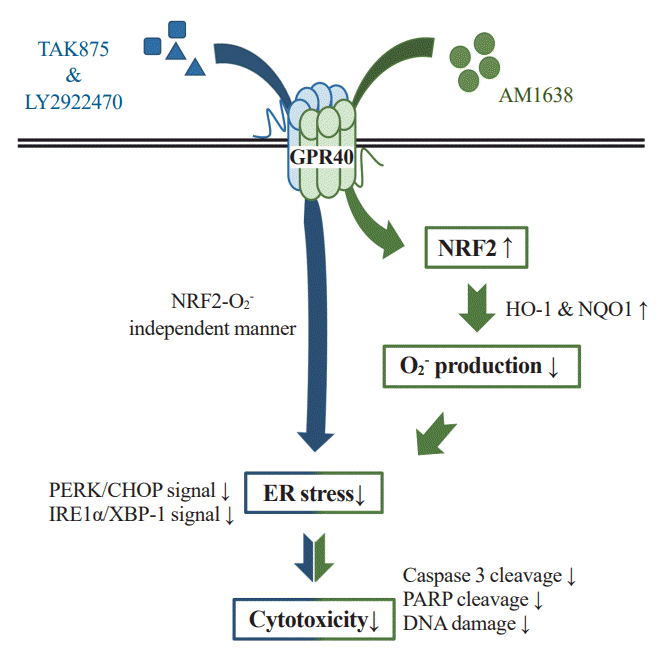
We obtained the following results in vascular endothelial cells: (1) AM1638, a GPR40-full agonist, increased the expression of antioxidant molecules (HO-1 and NQO-1); (2) AM1638 blocked palmitate-mediated superoxide production and ER stress; (3) AM1638 attenuated palmitate-induced cytotoxicity; (4) all the AM1638-mediated beneficial effects against palmitate were canceled by treatment with ML385, an NRF2 inhibitor; however, (5) TAK875 and LY2922470, which are GPR40-partial agonists, reduced palmitate-mediated ER stress and cytotoxicity without NRF2 activation.
GPR40 has two allosteric sites recognized by partial and full agonists. TAK875, a partial agonist, binds a region between TM3 and TM4 and activates only the G protein subunit alpha q (Gαq)-calcium pathway [15]. Full agonists, AM1638 and compound 1, attach near the intracellular loop 2 (ICL2) region and activate both the Gαq/calcium and Gαs/cyclic adenosine monophosphate (cAMP) pathways [16]. Both partial and full agonists can increase insulin secretion by enhancing the intracellular calcium level in human islets [17]. However, the amount of incretins, such as glucagon-like peptide-1 and glucose-dependent insulinotropic polypeptide, released from murine colonic crypts was strongly enhanced only by full agonist treatment in a cAMP-dependent manner; incretin level was not increased by partial agonist treatment [15]. We also found that GPR40 acts differently in vascular endothelial cells depending on the type of agonist. AM1638 significantly enhanced NRF2 translocation to the nucleus and the expression of its target genes, and those effects were not observed after GPR40 knockdown. On the other hand, TAK75 and LY2922470 did not induce NRF2-mediated signaling (Fig. 1A, B). Our best hypothesis to explain those differences is that each agonist induces a different GPR40 three-dimensional structure [15,16]. By examining the X-ray crystal structures between GPR40 and its agonists, Ho et al. [16] demonstrated that a full agonist preserved the structure of ICL2, which can interact with Gαs, while partial agonists induced ICL2 disorder.
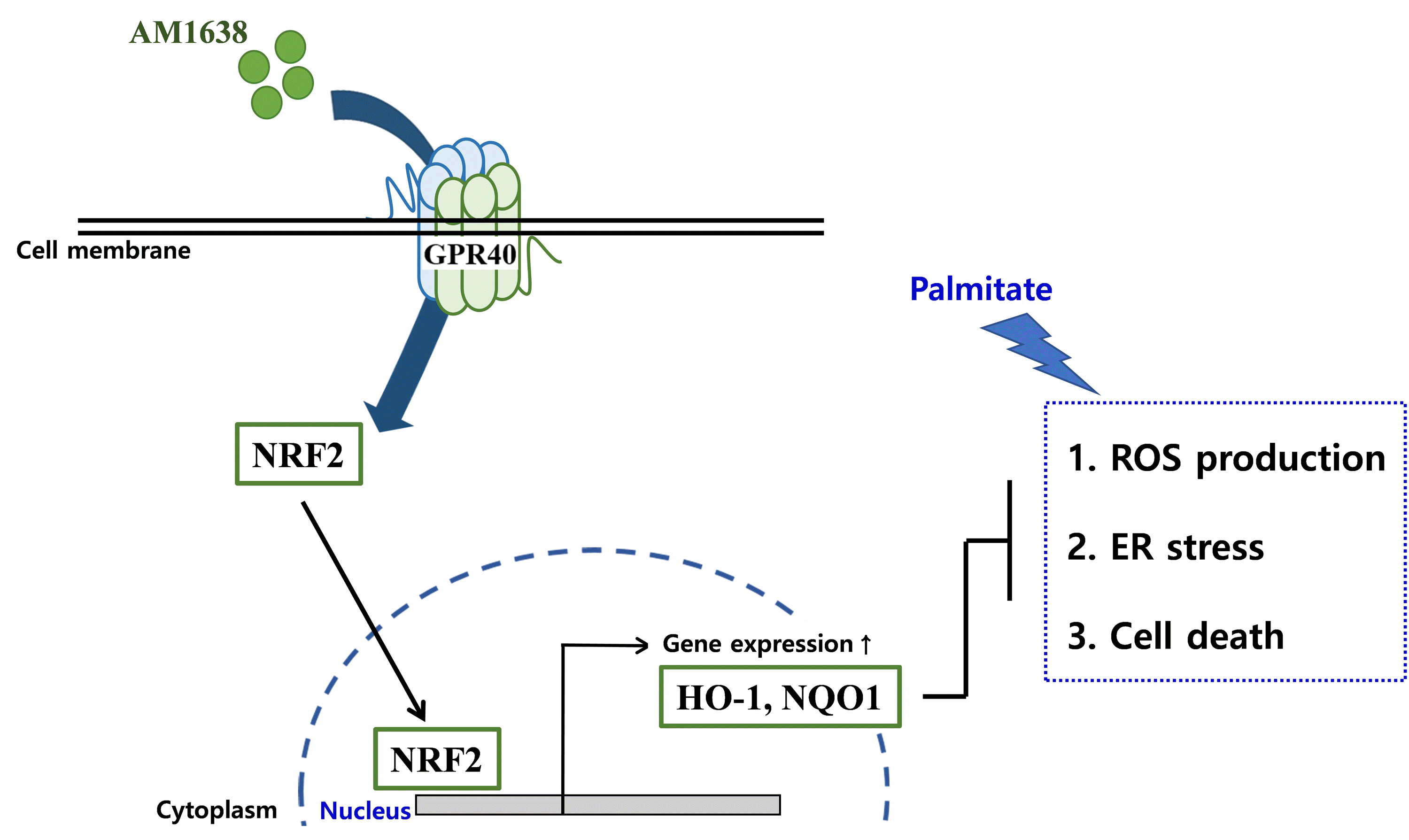
NRF2 is a regulator of the cellular defense system against oxidative stress. NRF2 translocated to the nucleus binds the antioxidant response element regions of DNA and initiates the transcription of antioxidant genes such as HO-1 [18]. Increased HO-1 generates biliverdin and carbon monoxide via heme degradation and enhances the expression of ferritin, which leads to anti-oxidative and anti-inflammatory responses [19]. HO-1 was more highly expressed in patients with atherosclerosis than in heathy people and in vulnerable plaques than in stable plaques [20,21]. Such upregulation has been considered a defense mechanism to inhibit the development of atherosclerosis because HO-1 showed anti-apoptotic and anti-thrombotic properties in the internal lumen of vascular vessels [22]. Therefore, we selected the NRF2-HO-1 axis as a good target for improving endothelial dysfunction. In our experiment, AM1638 treatment significantly increased the expression of HO-1 and NQO1, another NRF2 target gene, in HUVECs (Fig. 1B). AM1638 meaningfully reduced superoxide production and improved cell viability under palmitate treatment (Figs. 2A, 4B). Those beneficial effects were not observed after treatment with ML385, an NRF2 inhibitor (Figs. 2C, 4D). ML385 interacts to NRF2 and attenuates the binding ability of NRF2 to DNA [23]. Therefore, we suggest that GPR40-full agonists could be developed as novel therapeutics for vascular diseases such as atherosclerosis, as well as diabetes.
ER stress is involved with a prolonged and severe unfolded protein response induced by various metabolic stimuli, such as obesity, hyperglycemia, and ROS, that can lead to cytotoxicity and inflammation [24]. Therefore, endothelial ER stress is considered a major factor in the development of CVDs such as atherosclerosis and hypertension [25]. ER stress was increased more in unstable angina pectoris than in stable angina pectoris and created plaque vulnerability [26]. Treatment with 4-phenylbutyric acid, which inhibits ER stress, improved vasoconstriction and the media-to-lumen ratio and lowered blood pressure in hypertensive rats [27]. In this study, we experimented to learn whether GPR40 agonists reduced endothelial ER stress. Under palmitate treatment, AM1638 significantly inhibited ER stress–mediated molecular events such as the PERK-CHOP and IRE1α-XBP-1 pathways in HUVECs (Fig. 3B, C), and those effects were not observed after treatment with an NRF2 inhibitor (Fig. 3E, F). Likewise, cell viability improved after treatment with AM1638 in an NRF2-dependent manner (Fig. 4B, D). We expected that TAK875 and LY2922470 would not affect endothelial ER stress and cytotoxicity because they do not activate the NRF2/HO-1 axis and thus do not reduce palmitate-mediated superoxide production (Figs. 1B, 2A). Contrary to our expectation, TAK875 and LY292247 did decrease cytotoxicity by reducing ER stress in palmitate-treated HUVECs (Fig. 3B, C). Our results suggest that GPR40 might mediate beneficial actions other than NRF2/ROS signaling after treatment with a partial agonist. Nonetheless, the endothelial protective actions were greater after treatment with a full agonist than with a partial agonist (Fig. 4B). Based on our and previous results, we predict that a full agonist will be a better drug for correcting endothelial dysfunction than a partial agonist because a full agonist can inhibit endothelial ROS production (Fig. 2A, B) and increase the secretion of incretins [15], which are established therapeutic targets for CVDs [28].
Although clinical trials of TAK875 failed due to liver toxicity [29], GPR40 agonists still receive attention as anti-hyperglycemic agents. We describe here that, in HUVECs, GPR40 agonists can regulate palmitate-induced events differently depending on the type of agonist. GPR40-full agonists activated the NRF2/HO-1 axis, producing antioxidant and anti-apoptotic responses, whereas GPR40-partial agonists affected cell survival in an NRF2/HO-1 independent manner (Fig. 6). We thus propose that GPR40-mediated allosteric agonism could be an important therapeutic option for correcting endothelial dysfunction.
 XML Download
XML Download