

 PDF
PDF Citation
Citation Print
Print



Highlights
A PHEX variant (c.1483-1G>C) was found in an adult female patient by massively parallel sequencing. This finding helps us to understand the genotype-phenotype relationship and highlights the importance of genetic testing.
Introduction
X-linked hypophosphatemic rickets (XLH; OMIM number 307800) is an X-linked, dominantly inherited disorder characterized by defects in renal phosphate transport leading to phosphate wasting and hypophosphatemia. The incidence of XHL is 1 per 20,000 live births [1]. X-linked inheritance is the most common form of heritable hypophosphatemic rickets. The main clinical features of XLH are skeletal deformities, short stature, osteomalacia, dental abscess, and bone pain [1]. XLH is caused by a mutation in the PHEX gene located at Xp22.11. The PHEX gene consists of 22 exons, exhibits many similarities to the endopeptidase gene family, and functions as a phosphate-regulating endopepetidase [2,3]. The natural course of this disease is accumulation of unmineralized osteoids, leading to osteomalacia [4]. However, until recently, this disease was diagnosed predominantly based on clinical findings rather than genetic confirmation. Therefore, genetic characteristics of XLH are not fully understood. In this report, we describe a 25-year-old female Korean patient diagnosed with XLH harboring a rare pathogenic PHEX variant.
Case report
A 25-year-old female patient came to our clinic for genetic counseling regarding her presumed genetic disease and pregnancy. At the age of 1, due to delayed development, she was suspected of having developmental dysplasia of the hip or cerebral palsy. However, at 9 years, she was diagnosed with vitamin D-resistant rickets in another hospital. When she turned 10, due to her short stature, waddling gait, and bowing deformities of both legs, she underwent epiphysiodesis of both proximal tibias. Five years later, she had reached normal growth and sufficient bone mineral density for her age. When she visited our clinic for genetic counseling, her height and weight were 160 cm and 57 kg, respectively. Both of her parents were of normal height, and she reported no family history of rickets. Additionally, she did not have history of dental abscess in her childhood.
When she presented to our clinic, initial laboratory results showed normal serum calcium (9.7 mg/dL; reference value, 8.2–10.5 mg/dL), normal phosphorus (3.0 mg/dL; reference value, 2.5–4.5 mg/dL), high intact parathyroid hormone (PTH) (120.3 ng/L; reference value, 12–72 ng/L), low 25-OH vitamin D (18.7 ng/dL; reference value, 30–100 ng/L), normal 1,-25-(OH)2-vitamin D3 (25.81 ng/L; reference value, 19.6–54.3 ng/L), and high alkaline phosphatase (ALP) (123 U/L; reference value, 30–120 U/L). Serum fibroblast growth factor 23 was unchecked. A urine specimen showed normal random calcium to creatinine ratio (0.08; reference range <0.14). Laboratory findings were different from those of a typical XLH patient because she was on self-prescribed oral phosphorus and vitamin D. On physical examination, the patient was alert, with no signs of acute illness. After her endocrinologist increased the doses of phosphorus and calcium, her follow-up serum results showed normal calcium (9.7 mg/dL; reference value, 8.2–10.5 mg/dL), normal phosphorus (2.5 mg/dL; reference value, 2.5–4.5 mg/dL), high iPTH (86.1 ng/L; reference value, 12–72 ng/L), normal 25-OH vitamin D (30.4 ng/dL; reference value, 30–100 ng/L), normal 1,-25-(OH)2-vitamin D3, and normal ALP (108 U/L; reference value, 30–120 U/L). Follow-up urine test showed normal random calcium to creatinine ratio (0.02; reference range, <0.14). Dual energy X-ray absorptiometry measurement showed mean bone mineral density in right hip, left hip, and lumbar spine (L1 to L4) was 0.853 g/cm2 (T-score of 0), 0.998 g/cm2 (T-score of 1.3), and 1.284 g/cm² (T-score of 2.4), respectively.
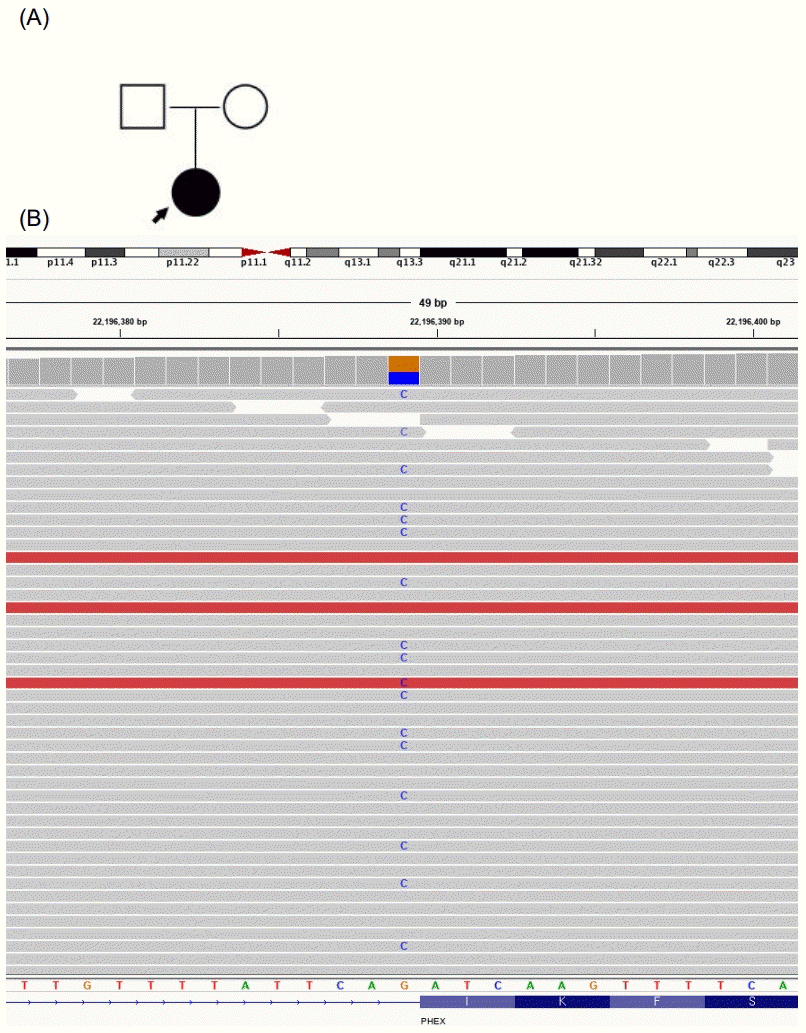
She was concerned whether her condition might affect her future child and wished to confirm the causative gene behind the condition. A multigene panel associated with skeletal dysplasia (composed of the following 30 genes: ALPL, ARSES, COL10A1, COL1A1, COL1A2, COL2A1, COL9A1, COL9A2, COL9A3, COMP, CRTAP, CTSK, EBP, EXT1, EXT2, FGF23, FGFR1, FGFR2, FGFR3, FLNB, GNPAT, MATN3, P3H1, PEX7, PHEX, RUNK2, SLC26A2, SLC34A3, TGFB1, and TRPS1) was conducted. A splicing site mutation in exon 14 of PHEX, a heterozygote of the c.1483-1G>C variation (NM_000444.5), was found (Fig. 1), as had been reported in another study [5]. The finding was a pathogenic variant of the PHEX gene according to the American College of Medical Genetics and Genomics and the Association for Molecular Pathology 2015 guidelines. The patient had a null variant in a gene where loss of function is a known mechanism of disease (PVS1) and a de novo mutation (PS2), the variant was absent from controls in Exome Sequencing Project (PM2), and her phenotype was highly specific for a disease with a single genetic etiology (PP4) [6]. In splicing prediction tools, both SPF (https://www.genomnis.com/) and varSEAK (https://varseak.bio/) predicted c.1483-1G>C to cause a splicing defect. Both of her parents were negative by direct sequencing analysis of the PHEX gene. In addition, short tandem repeat PCR was performed to confirm paternity. Hence, the patient was diagnosed with de novo XLH. There was a possibility of X-linked recessive disease because none of her family members presented history of rickets. XLH caused by a c.1483-1G>C variant has only been reported once, in 2016 in two Chinese family members whose main symptoms were growth retardation, short stature, and genu varum [5].
Discussion
PHEX gene mutation is the main cause of XLH. This gene contains 22 exons encoding a transmembrane endopeptidase of 749 amino acids. This protein is expressed mainly in osteoblasts, osteocytes, and odontoblasts [7]. A 6.6 kb mRNA transcript of the PHEX gene contains 35% (2,247 bp) coding sequence and 65% untranslated regions [1]. As of April 2020, 407 PHEX gene variants causing XLH were listed in the Human Gene Mutation Database [8]. The most common mutation type is missense or nonsense, comprising up to 37.3% (152 of 407) of total mutations. In addition to an X-linked pattern, autosomal dominant and autosomal recessive forms of hypophosphatemic rickets have been reported, the latter with a much lower incidence [7]. Sporadic cases are common, and many subsequently transmit the phenotype in an X-linked dominant manner [1]. Mutation detection rates are significantly lower in sporadic cases (23% to 47%) compared to the cases with family history [9]. In an Italian study, 12 of 19 patients with PHEX mutation had sporadic pattern of inheritance [7]. The explanation for this fluctuating incidence of de novo XLH could be that the parents of all the patients were not tested for the genetic mutations.
There is wide variability in XLH phenotypes, ranging from mild hypophosphatemia to severe bone deformities [1]. Several studies have investigated the correlation between phenotypical severity and genetic profile. According to Holm et al. [1], no correlation was found between disease severity and type or location of the mutation in analysis of 31 hypophosphatemic rickets patients with PHEX mutation. Similarly, in a Korean study on 17 children, no correlation was noted between genotype or phenotype with any gene dosage effect [9]. A weak correlation was supported by an Italian study involving 26 patients with hereditary rickets [7]. However, among patients with a family history of hypophosphatemic rickets, a trend toward more severe skeletal disease was observed in patients with truncating mutations such as nonsense, deletion, insertion, or splice site mutations [1]. Additionally, skeletal disease was more severe with C-terminal mutations than with those of the N-terminal [10]. This correlates with our patient, whose c.1483-1G>C variant is located relatively close to an end of the gene. Although hearing defect, dental disease, and increased head length were not observed in our patient, these clinical manifestations correlated in the beginning and terminal fragments of the gene [9]. In the second year of life, affected children usually exhibit bowing deformities of the lower extremities, become resistant to physiologic doses of vitamin D, and show growth retardation, as in our patient. Later in life, the patients can develop dental abscesses, extraskeletal ossification, osteomalacia, and bone pain [11].
In conclusion, we identified a pathogenic PHEX splice site mutation in a Korean female patient diagnosed with sporadic XLH. The relationship between this variant and her phenotype correlated with previous studies. Moreover, using genetic analysis in diagnosing hereditary hypophosphatemic rickets is important in terms of precise clinical management and patient understanding though genetic counseling. Additional clinical and molecular studies involving patients with XLH are needed to determine the relationship between genotypic and phenotypic expression and to elucidate the pathophysiology of XLH.
 XML Download
XML Download