

 PDF
PDF Citation
Citation Print
Print



Introduction
Type 2 diabetes (T2D) is a major health problem that has reached epidemic proportions around the world. It is correlated with several metabolic and cardiovascular complications in both adults and children [1,2].
Insulin resistance (IR) has been identified as a cardinal trigger of impaired glucose metabolism, T2D, and cardiovascular diseases [3,4]. The term 'insulin resistance' refers to a reduced glucose uptake in response to physiological insulin levels. It is a common feature of childhood obesity and is quite detectable in children and adolescents [5].
Beta-cell dysfunction and IR are key defects contributing to the development of T2D [6,7]. Therefore, understanding the mechanisms correlated with the decline of beta-cell function and with IR is crucial for controlling, preventing, and treating T2D, particularly in youth.
The aim of this review is to discuss the current knowledge on IR and T2D, which can provide interesting opportunities and stimulating challenges for the development of new preventative approaches and therapeutic strategies for children and adolescents with T2D.
T2D in children
1. Dimension of the problem
Over the past 20 years, there has been an alarming increase of T2D in youth, concomitant with the rise of obesity. Indeed, current data indicate an incidence of ~5,000 new cases per year of pediatric T2D in the United States [8]. The Centers for Disease Control and Prevention and the National Institutes of Health performed a population-based observational study of diabetes in youth also demonstrating an increasing prevalence of T2D in United States between 2001 and 2009. In particular, the overall prevalence of T2D in youth was reported as 0.34 per 1,000 in 2001 and 0.46 per 1,000 in 2009, indicating a relevant increase of 35% [9]. Suggesting a 2.3% annual increase, the SEARCH database showed that in subjects under 20 years of age, the prevalence of T2D will quadruple in 40 years [10].
Similar trends have also been reported in many European countries, although there is a significantly lower incidence rate of T2D. In particular, Candler et al. [11] showed an incidence of T2D of 0.72/100,000 per year in British children and adolescents. Similarly, Schober et al. [12] described the incidence of T2D at 0.29 cases per 100,000 per year between 1999 and 2007 in Austrian children and adolescents.
In Korea the average crude incidence rate of T2D was reported as 0.75/100,000 from 2001 to 2010, with an increasing trend over the 10-year period [13]. Similarly, Chinese studies have detected an increased incidence of pediatric diabetes in different geographical areas [14]; they have also shown a T2D prevalence of 2.52/100,000 in the southwest, 3.77/100,000 in the east and 15.64/100,000 in the north [15]. Similarly, in New Zealand the incidence of T2D in children younger than 15 years of age has increased gradually at 5% per year over the last 21 years; the overall annual incidence of T2D in children under 15 years of age was detected as 1.5/100,000 [16,17]. In 2006–2008, the incidence of childhood-onset T2D in Canada was assessed at 1.54 cases per 100,000 per year [18].
Interestingly, it was also documented that minority races and ethnicities have the highest incidence and prevalence of T2D, probably due to genetics and hormonal factors, environmental influences, different quality of life, and access to health care. The results of a study from UK showed that Black individuals had 3.9 cases of T2D per 100,000 person-years, while South Asian and Caucasian individuals had rates of 1.25 and 0.35 per 100,000 person-years, respectively [19]. In the U.S. population, Pavkov et al. [20] described that the highest incidence of T2D was calculated in 4- to 15-year-old Pima Indians (330 per 100,000 person/yr).
Thus, T2D disproportionately involves youth of ethnic and racial minorities, demonstrating substantial differences in insulin secretion and sensitivity that might increase the risk of T2D. However, a complex of psychosocial and cultural environments may also influence the onset of T2D in these ethnic and racial minorities, where it may be difficult to achieve healthy lifestyle modifications and self-management actions.
2. Pathophysiology
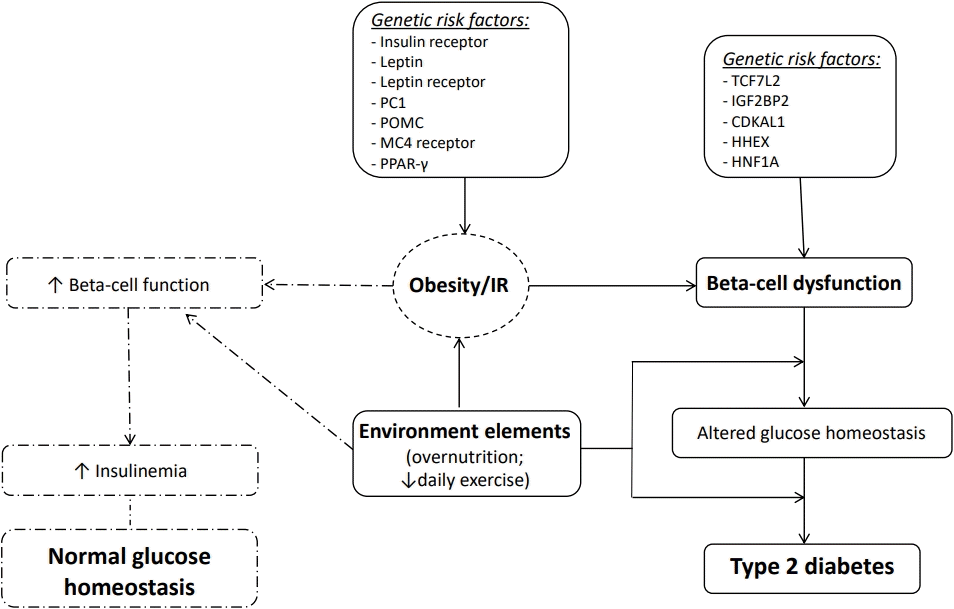
The pathophysiology of T2D in children is analogous to, but more complex than, the disease process in adults [21]. Indeed, many authors suggest that the rate of beta-cell dysfunction occurs more rapidly than in adults [22]. T2D is a complex and multifactorial disease, characterized by the association between genetic, epigenetic, and environmental factors (Fig. 1).
Certainly, obesity is considered the most important risk factor for the development of T2D. However, not all obese children develop T2D; indeed, T2D can be diagnosed in children with a relatively lower body mass index percentile compared to their peers. Thus, many other factors are certainly involved in the pathogenesis of T2D [23] (Table 1). More than 40 genes may increase the risk of T2D in adults, with the most relevant diabetes susceptibility gene identified as transcription factor 7-like 2 (TCF7L2); this gene appears to increase diabetes risk 1.7-fold [24]. Several other studies have also investigated the impact of genetic risk in healthy youth and children with prediabetes and/or T2D. In 2010, Dupuis et al. [25] performed the MAGIC (Meta-Analyses of Glucose and Insulin-related traits Consortium) study, which described a large-scale meta-analysis of genome-wide data for investigation of genetic correlations with fasting glycemic traits in Caucasian European children. The study showed that the largest effects sizes were detected for risk alleles in Glucokinase (GCK), Glucose-6-Phosphatase Catalytic Subunit 2 (G6PC2) and Melatonin Receptor 1B (MTNR1B). Interestingly, an additional 5 loci (GLIS3, PROX1, SLC2A2, ADCY5, and CRY2) a were also correlated with higher fasting glucose in youth [26]. Giannini et al. [27] evaluated a multiethnic cohort of obese children and reported that the co-occurrence of risk alleles in or near 5 genes modulating insulin secretion (TCF7L2 rs7903146, IGF2BP2 rs4402960, CDKAL1 rs7754840, HHEX rs1111875, and HNF1A rs1169288) was associated with a higher risk of prediabetes/T2D in obese children and adolescents. Recently, Cropano et al. [28] explored the mechanisms by which the TCF7L2 rs7903146 risk allele confers susceptibility to impaired glucose tolerance (IGT) or T2D in obese adolescents. This study showed that this variant can impair beta-cell function and hepatic insulin sensitivity, thus predicting the development of IGT/T2D over time. Therefore, beta-cell dysfunction is a crucial determinant for T2D associated with IR. However, the existing correlation between beta-cell dysfunction and IR remains highly complex.
Adequate glycemic control is regulated by a balance between insulin secretion from the beta-cells and sensitivity to insulin in adipose tissue, skeletal muscle and liver [29]. However, reduced insulin sensitivity has been documented during puberty and in obese children, and this is correlated with high insulin secretion levels. Nonetheless, when beta-cells are unable to produce sufficient insulin to compensate for IR, alterations in glucose homeostasis occur and progress to prediabetes and T2D as beta-cell function is further reduced [30].
Many authors have performed cross-sectional and longitudinal studies in children and adolescents with obesity demonstrating that, as in adults, beta-cell failure with progressive reduction of insulin secretion relative to impaired insulin sensitivity paves the way for prediabetes and T2D in high-risk youth [31,32]. In particular, many studies have reported that metabolic alterations, such as glucolipotoxicity and consequent production of reactive oxygen species and reactive nitrogen species, could interfere with beta-cell function [33,34]. The resulting lower adenosine triphosphate synthesis, associated with insufficient antioxidant factors, is also involved in beta-cell failure.
3. Risk factors, screening, and diagnosis
Many risk factors are involved in youth-onset T2D including genetic and epigenetic elements, pubertal stage, first- or second-degree relatives with T2D, low socioeconomic status, and being the offspring of a pregnancy characterized by gestational diabetes mellitus (GDM). Indeed, during the first decade of life, it is possible to detect metabolic evidence of genetic susceptibility to T2D by documenting compromised insulin secretion and insulin sensitivity in otherwise healthy children with a positive family history [35].
However, it is during the pubertal period that T2D can be detected more frequently, when physiologic and transient IR occurs (insulin sensitivity declines by 25%–30%) [36]; therefore, the diagnosis of T2D in youth is usually reported at a mean age of 14 years [37].
In addition, maternal obesity and GDM seem to play a relevant role in promoting obesity and T2D in offspring. In 2016, Chernausek et al. [38] reported that maternal diabetes prior to or during pregnancy was correlated with worse glycemic homeostasis and beta-cell dysfunction in children, supporting the hypothesis that fetal exposure to impaired glycemic profiles may have late effects during life. Similarly, Dabelea et al. [39] reported that youth with T2D were more likely to have been exposed to maternal diabetes or obesity in utero than nondiabetic control youngsters. Therefore, prevention strategies may need to target the increasing number of pregnancies characterized by obesity and diabetes in addition to childhood obesity itself, especially as GDM seems to correlate with a younger age of T2D onset.
However, the risk of developing T2D also includes potentially modifiable risk factors such as obesity, lack of physical activity, excess nutritional intake, and sedentary life.
Indeed, risk-based screening for T2D should include children and adolescents after the onset of puberty or after 10 years of age, whichever occurs earlier, in overweight (body mass index [BMI] ≥85th percentile) or obese (BMI ≥95th percentile) children and youth that have one or more risk factors for diabetes (Table 2). It has also been reported that increasing BMI could be a predictor of deteriorating glycemia and progression to T2D. Therefore, these youngsters should be more frequently screened for T2D.
Screening with fasting glucose, oral glucose tolerance test (OGTT), or hemoglobin A1c (HbA1c) is an adequate approach, but should be associated with a thorough clinical evaluation. If tests are normal, they should be repeated at a minimum of 3-year intervals, or more frequently if BMI is increasing [40]. In at-risk obese children and adolescents, the presence of type 1 diabetes-associated beta-cell autoantibodies and genetic screening for monogenic diabetes should also been performed when appropriate. The diagnostic criteria for T2D are the same for young patients and adults (Table 3) [40].
Insulin action and resistance in T2D
1. Insulin signaling and IR
It is well known that insulin is a potent anabolic hormone produced by pancreatic beta-cells and its principal metabolic actions are to stimulate glucose uptake in adipocytes and skeletal muscles, inducing glycogen synthesis in skeletal muscles, inhibiting hepatic glucose output, and suppressing lipolysis in adipocytes.
Normally, the glucose transporter 2 modulates glucose secretion in hepatic tissue, whereas the insulin-sensitive glucose transporter 4 (GLUT4) regulates glucose uptake in fat and muscle [41]. The insulin signaling pathway stimulates a key protein kinase Akt to achieve adequate blood glucose homeostasis [42]. This Akt protein kinase is fundamental for insulin’s influence on the mechanisms that regulate systemic glucose levels, including glucose concentration in adipocytes and muscles, prevention of hepatic gluconeogenesis, and induction of hepatic lipogenesis [43]. Insulin signaling can also promote lipid storage in adipocytes by inducing triacylglycerol synthesis and inhibiting lipolysis. Triglycerides are stored in lipid droplets, which are composed by lipid droplet proteins such as perilipin [43]. In addition, insulin plays a relevant role in vascular tissue, which can have either protective or deleterious effects. Indeed, insulin's action is strictly correlated with the production of endothelial nitric oxide synthase; however, it also promotes vascular smooth muscle cell proliferation, proinflammatory activity, and systemic vasoconstriction [44].
IR is a state in which insulin has a subnormal biological effect. In particular, it is characterized by a reduced capacity of insulin to induce the use of glucose by muscles and adipose tissue and inhibit hepatic glucose production and output [45]. IR plays a crucial role in skeletal muscles, in which it is able to reduce glycogen synthesis and protein catabolism; it also inhibits lipoprotein lipase activity in adipocytes, thus leading to elevated secretion of free fatty acids and inflammatory cytokines such as tumor necrosis factor alpha (TNF-α), interleukin (IL)-6 and leptin. Moreover, it causes altered glucose output and fatty-acid metabolism and leads to elevated triglyceride concentration due to very low density lipoprotein secretion from hepatic tissue [46]. In addition, IR influences endothelial cell function by interfering with the production of nitric oxide and stimulating the release of pro-coagulant factors that determine platelet aggregation. IR is also a key player in the pathogenesis of metabolic diseases like T2D, nonalcoholic fatty liver disease, and cardiovascular disease [47].
2. Methods of measurement and diagnosis of IR
IR is diagnosed frequently in children, but it is rarely measured directly in the pediatric population. The hyperinsulinemic-euglycemic clamp (HEC) is the gold standard test for the evaluation of IR in children [48]. It is a direct measure of the insulin-stimulated glucose disposition correlated with a given degree of hyperinsulinemia [5]. This technique has been able to recognize specific causes of IR in childhood and during adolescence. However, this test is lengthy (2–3 hours), requires expertise of operators for constant adjustment of the glucose infusion rate, and carries a risk of hyperglycemia [49,50].
Indeed, in the last year, numerous surrogate measures have been developed. Some of these methods are based on measurements of fasting blood samples, whereas others require measurements derived from an OGTT, which is frequently used, or an intravenous glucose tolerance test, which is not suitable for daily practice [51]. However, their clinical use is limited because of the absence of reference values for normal and impaired insulin sensitivity; for clinical purposes, the Homeostatic Model Assessment for Insulin Resistance (HOMA-IR), the Quantitative Insulin Sensitivity Check Index (QUICKI), and Matsuda index are usually used, while the HES, Belfiore, McAuley, Cederholm, Avignon and Stumvoll indices are suitable for epidemiological/ research purposes [52] (Table 4).
Fasting plasma insulin (FPI), HOMA-IR, and QUICKI are the most common surrogate markers which correlate favorably and have been validated in childhood with the HEC [53,54]. The FPI is generally used as surrogate index of insulin sensitivity and evaluates insulin concentration after a fasting period, but it represents an index of liver insulin metabolism; it evaluates only a state of hyperinsulinemia and does not correlate very well with IR in children and adolescents [55].
HOMA-IR is the most commonly used method in daily practice and evaluates IR through fasting glucose and insulin levels. HOMA-IR was found to be much more reliable than fasting glucose/insulin ratio and QUICKI in determining IR in obese children [56]. Indeed, HOMA-IR is a frequently-used parameter in clinical research [57,58].
The QUICKI index is similar to HOMA, but it is more complicated because it uses the reciprocal of the logarithm of both insulin and glucose to account for the skewed distribution of fasting insulin values. The log-transformation leads to greater accuracy than HOMA [59]. Accordingly, the QUICKI and HOMA indexes are inversely related and they present the same limitations.
Furthermore, the triglyceride to high density lipoprotein cholesterol ratio (TG/HDL-C) is useful markers of IR because low triglyceride concentrations are usually associated with increased insulin sensitivity, especially in Caucasian populations [60].
The McAuley Index is another surrogate index which has a higher sensitivity and similar specificity to fasting insulin alone when evaluating IR, but to date it has been used only in adults [61]. Another recently proposed index is the variation of sex hormone binding globulin as a biomarker of IR, but it is not widely used [62].
Other markers of IR calculated during OGTT have been described recently by Monzillo and Hamdy [63] and Matsuda and DeFronzo [64]; they are considered more reliable than fasting sample indices, because they are able to evaluate a postload interaction between glucose and insulin. The ISI0,120, defined as the Matsuda index, was adapted from the SI index and developed by Cederholm and Wibell [65]. This formula uses only the 0- and 120-minute glucose and insulin values obtained from the OGTT (excluding the 30- and 60-minute values included in the SI index) to improve precision and calculate the glucose uptake rate in peripheral tissues. However, there is no consensus yet on which method and cutoff value has more advantages in terms of reproducibility, sensitivity, specificity, and simplicity [66].
Therefore, all methods have been used concurrently, which impedes comparisons of incidence and prevalence rates of IR between populations and countries as well as studies of these rates over time [51].
3. New indexes of IR
Rivera et al. [67] studied alterations in circulating proinflammatory and anti-inflammatory cytokines (TNF-α, IL-1β, IL-4, IL-6, and IL-10), chemokines (stromal cell-derived factor-1alpha, monocyte chemoattractant protein 1 [MCP-1], eotaxin, and fractalkine) and growth factors (brain-derived neurotrophic factor, platelet-derived growth factor [PDGF-BB] and insulin-like growth factor-1 [IGF-1]) in obese children with IR and evaluated the usefulness of these as a diagnostic tool. Obesity-mediated IR determines adipose tissue inflammation, accumulation of macrophages, production of pro-inflammatory cytokines, and TNF-α and IL-1β [68,69]. In addition, IR is associated with increased production of chemokines, such as MCP-1 and eotaxin, which are expressed in preadipocytes, as well growth factors such as PDGF-BB. Reduced circulating levels of PDGF-BB (a pro-fibrotic growth factor that plays an important role in the development of adipose tissue vascularization) was able to distinguish IR and non-IR in prepubertal children with obesity; the association of several biomarkers, such as leptin, TG/HDL, IGF-1, TNF-α, MCP1, and PDGF-BB can be considered as relevant diagnostic tools to detect IR in prepubertal children with obesity. In particular, changes in circulating concentrations of TNF-α, MCP1, and PDGF-BB were consistently correlated with IR in obese children [67].
Da Silva et al. [70] examined the accuracy of HOMA-Adiponectin in detecting IR based on the hyperglycemic clamp technique in adolescents. Adiponectin increases fatty-acid oxidation in muscle and liver via activation of AMP-activated protein kinase and peroxisome proliferator-activated receptor alpha mediated by a specific receptor [71]. The correlation between adiponectin and insulin sensitivity in adolescents has also been reported [72,73].
Kang et al. studied another surrogate marker, the "triglycerides/glucose index" (TyG) calculated as ln [triglycerides (mg dL-1) ×fasting glucose (mg dL-1)/2]. This index was correlated with HOMA-IR and showed a strong positive association with TG/HDL-C. The cutoff of the TyG index proposed for the diagnosis of IR was 8.18; other authors propose values between 3.7 and 4.5. regardless, the TyG index can be a simple, cost-effective surrogate marker of IR among adolescents compared with HOMA-IR [74-77].
Obesity, IR, and T2D: a possible unifying mechanism
Identifying a single pathway to explain the link between obesity, IR, and T2D would be ideal. Certainly, beta-cell dysfunction, which determines impaired insulin release, plays a crucial role in the pathophysiology of T2D [78]. In particular, impaired insulin output could interfere with physiological glucose uptake in adipose tissue and induce increased lipolysis and elevated nonesterified fatty-acid concentrations. Moreover, beta-cell dysfunction could also interfere with physiological insulin action in crucial brain regions, resulting in weight gain and aggravating IR [79].
IR triggers a rise in insulin need and normally stimulates beta-cell activity by increasing both beta-cell mass and insulin secretion, leading to hyperinsulinemia. However, in a vicious cycle, hyperinsulinemia can exacerbate the metabolic dysregulations that cause beta-cell failure and the development of T2D [80,81].
Accordingly, some studies in mice and in humans have induced IR by disrupting molecular insulin signaling in skeletal muscle, adipose tissue, and the liver, thus leading to hyperinsulinemia and diabetes. These studies have reported high levels of circulating insulin in enrolled subjects with consequent onset of diabetes [42,82].
The possible initiating pathways that could explain the correlations between obesity, IR, and hyperinsulinemia could include the activation of the transcription factor FOXO1 in the liver [83] and alteration of glucose transporter (GLUT4) translocation to the surface membrane in skeletal muscle [84]. The upregulation of FOXO1 can lead to improved conversion of incoming substrates to the liver to glucose; on the contrary, reduced GLUT4 concentrations in muscle would impair glucose uptake from the circulation [85]. Indeed, it was reported that in mice with obesity, FOXO1 was upregulated and these mice became insensitive to insulin regulation [86]; how obesity and overnutrition could interfere with FOXO1 is still not completely understood [87]. Many authors have hypothesized that the molecular mechanisms described above could appear exaggerated in T2D, where beta-cells cannot produce enough insulin to compensate the upregulation of FOXO1 [78].
However, other authors have speculated that hyperinsulinemia is the primary effect of a high-fat diet (HFD) and obesity, consequently stimulating beta-cells [88]. According to this hypothesis hyperinsulinemia determines IR, hyperglycemia, and hyperlipidemia. In particular, a HFD and overfeeding could determine insulin release from pancreatic beta-cells, causing hyperinsulinemia [89,90].
In any case, more studies are needed to evaluate the molecular mechanisms that modulate pathways which in turn may induce IR, hyperinsulinemia, and beta-cell dysfunction in T2D patients. A growing number of studies in adults [91,92] have recently identified many inflammatory markers that could explain these molecular patterns. Although the role of these markers could not be confirmed in children, other inflammatory markers have been studied as potential links between IR and T2D [93]. Interestingly, these results support the hypothesis that systemic inflammation is one possible molecular pathway that could link obesity, IR, and T2D in youth.
Unfortunately, none of these potential new biomarkers have proven prognostic value compared to traditional assessments, i.e., measurement of HbA1c. Therefore, their value for routine clinical purposes remains to be elucidated.
Concluding remarks
Beta-cell dysfunction and consequently insufficient insulin secretion characterize the advanced state of diabetes. IR plays a crucial role in the pathogenesis of T2D, hypertension, and cardiovascular risk. Thus, understanding the molecular pathways of IR is fundamental to guide the development of future therapeutic approaches for adults and children with T2D. HFD and obesity trigger IR and strongly modulate the onset of T2D in youth. Therefore, beta-cell preservation through healthy lifestyle, proper nutrition, daily exercise, and only if necessary, therapeutic interventions, become fundamental for the treatment and prevention of T2D and its cardio-metabolic complications. Indeed, understanding the complex pathways that compose the molecular mechanisms of T2D is crucial to identify accurate and personalized therapeutic targets for treating IR and preventing T2D.
 XML Download
XML Download