

 PDF
PDF Citation
Citation Print
Print



Introduction
Congenital hyperthyroidism, which is very rare, can be classified into 2 categories: autoimmune hyperthyroidism and non-autoimmune hyperthyroidism. Autoimmune hyperthyroidism accounts for most cases of congenital hyperthyroidism; in this disease, thyroid-stimulating antibodies (TSAbs) from a mother with Graves' disease cross the placenta and cause transient hyperthyroidism in the infant [1]. In contrast, non-autoimmune hyperthyroidism is caused by constitutive activation of the thyroid-stimulating hormone (TSH) receptor (TSHR) from an activating mutation in the TSHR gene [2]. Activating germline mutations in the TSHR gene are inherited in an autosomal dominant pattern or may appear sporadically; these are referred to as familial non-autoimmune autosomal dominant hyperthyroidism (FNAH) and persistent sporadic non-autoimmune hyperthyroidism (PSNAH), respectively. Although the symptoms of hyperthyroidism in patients with PSNAH manifest at relatively young ages, even during the fetal period, clinical symptoms of FNAH may manifest at various ages (from the neonatal period to 60 years), with symptoms ranging from mild to severe [3,4]. According to recent reports (http://www.tsh-receptor-mutation-database.org/), approximately 29 types of activating TSHR gene mutations causing FNAH have been reported through 2018.
In this report, we describe the second report of a p.Ala627Val mutation that was confirmed as a germline variant in the TSHR gene, in which the patient was diagnosed with familial non-autoimmune hyperthyroidism.
Go to : 
Case report
An 80-day-old boy presented with increased irritability compared with his twin sister. The patient was born as the younger twin through Cesarean section at 36 weeks and 6 days of gestation, and there were no perinatal problems. At the time of birth, his body weight was 3.06 kg (25th–50th percentile), height was 50 cm (75th–90th percentile), and head circumference was 34 cm (75th–90th percentile). No abnormal findings were noted on the newborn screening test conducted on day 3, and neonatal TSH (1.4 μU/mL; reference range, 0.0–12.0 μU/mL) and neonatal T4 (13.3 μg/dL; reference range, 5–22 μg/dL) were normal. On family history, the patient's mother had been taking methimazole for 12 years for Graves' disease. Therefore, the patient was scheduled for a follow-up thyroid function test one week later. However, the patient's parents did not attend the outpatient appointment. Instead, the patient presented at 80 days of age owing to increased irritability compared with his twin sister. On thyroid function test, although free T4 (FT4) was normal, TSH was suppressed (serum T3: 272.1 ng/dL [reference range, 80–200 ng/dL]; FT4: 2.08 ng/dL [reference range, 0.93–2.6 ng/dL]; and TSH: 0.0 μIU/mL [reference range, 0.6–5.6 μIU/mL]). Pulse, respiratory rate, and body temperature were within normal ranges. On physical examination, the patient was alert, with no signs of acute illnesses. There were no signs of enlarged thyroid, exophthalmos, or enlarged jugular vein, and the patient’s development was appropriate for 3 months of age. Tests of antithyroglobulin antibody, antithyroid peroxidase antibody, and anti-TSHR antibody were negative. However, since the patient’s mother had Graves' disease, the findings were thought to have resulted from transient autoimmune hyperthyroidism. Therefore, the patient was not started on methimazole and was instead scheduled for short-term regular follow-up. Although the patient remained asymptomatic, at 4 months of age, FT4 increased to 2.89 ng/dL and TSH remained suppressed at 0.01 μIU/mL. Thus, pharmacological treatment was initiated with 3-mg/day methimazole. However, one month after starting methimazole, a hypothyroid state was observed (FT4, 0.87 ng/dL; TSH, 12.1 μIU/mL), and the medication was discontinued. Thyroid function tests that were conducted every month showed elevated FT4 and suppressed TSH; therefore, methimazole at 3 mg/day was started again at 9 months of age (Table 1). Thyroid gland ultrasonography conducted at 9 months was normal, and the patient remained negative for TSAbs. Fig. 1 shows the patient's serum FT4 and TSH over 34 months. Because continued use of methimazole was needed to maintain normal thyroid function, we considered non-autoimmune hyperthyroidism due to TSHR gene mutation.
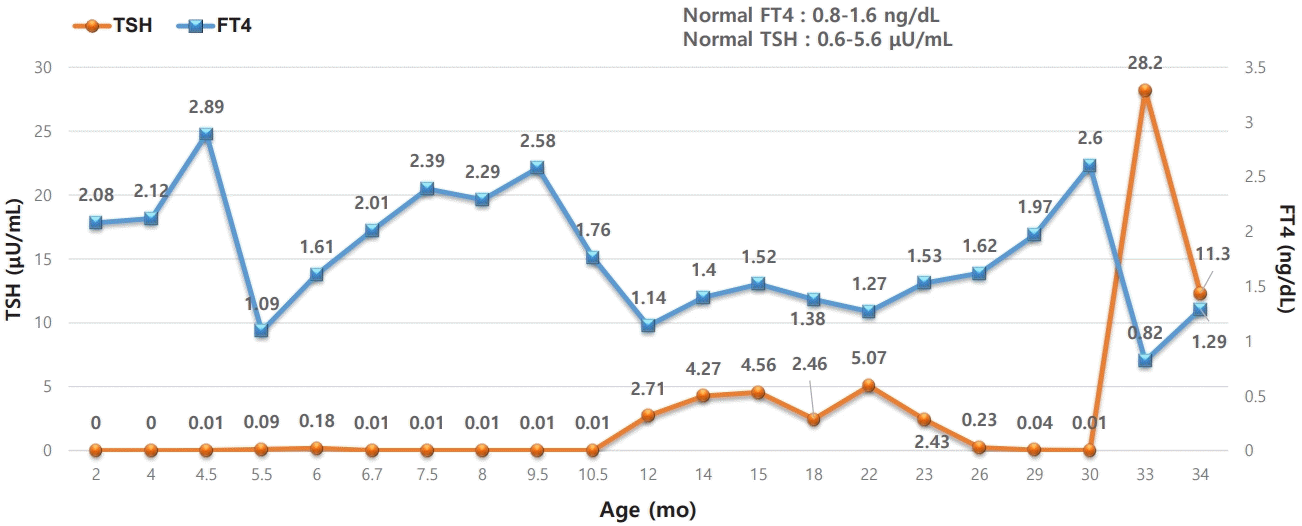
Table 1.
Results of thyroid function tests for the patient
![]()
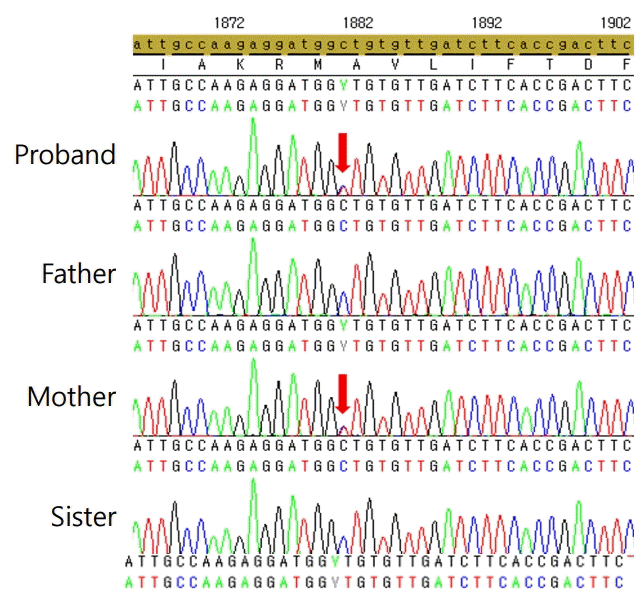
Based on our findings, we performed whole-exome sequencing (WES), and genomic DNA was extracted from a buccal swab sample from the proband. All exon regions of all human genes were captured using the SureSelect kit (Version C2; Agilent Technologies, Inc., Santa Clara, CA, USA). The captured regions of the genome were sequenced with a NovaSeq platform (Illumina, San Diego, CA, USA). Variant calling, annotation, and prioritization were performed as previously described [5]. The heterozygous missense variant c.1880C>T in TSHR had a valine instead of an alanine at codon 627 in exon 10. This variant has not been reported in large population cohorts [6]. In silico analysis predicted that the variant was disease-causing, according to REVEL [7]. In addition, the variant was located in an exonic hotspot where 9 pathogenic variants have been reported [8]. The variant was classified as likely pathogenic (evidence: PM1, PM2, PP2, PP3, and PP4) according to the recommendation of the American College of Medical Genetics and Genomics guidelines [9]. This variant was tested and confirmed via direct sequencing from the peripheral blood genomic DNA of the proband and his family members. This missense variant also was found in the patient's mother, who suffered from hyperthyroidism. The patient's variant was considered an inherited mutation from his mother. The variant was not found in the patient's father or twin sister (Fig. 2). FNAH caused by a c.1880C>T (p.Ala627Val) variant in the TSHR gene has been reported only once, in 2005 in a South Korean family in brothers and their mother [10]. Our patient is currently 2 years 10 months old and is maintaining normal thyroid function without notable side effects on regular use of methimazole 3 mg/day.
Go to :
Discussion
In this report, we describe a case of mild FNAH caused by A627V activating germline mutation in the TSHR gene; the patient's family was the second to be diagnosed worldwide with the A627V variant. However, the same variant has been reported as a somatic mutation in the TSHR gene in a patient that showed toxic thyroid nodules [11]. According to previous studies, the A627V mutation is located within the transmembrane helix, and TSHR activation owing to the alanine (GCT) to valine (GTT) substitution results in toxic thyroid nodules with hyperthyroidism [11]. The same variant was found in brothers with non-autoimmune hyperthyroidism, reported in 2005 [10]. The patients' mother also exhibited hyperthyroidism and was confirmed to have the same TSHR mutation as the brothers, satisfying the diagnosis of FNAH. Moreover, the patients did not show any symptoms other than sweating and palpable thyroid nodules, which were first palpated at 4 years of age [10]. The A627V variant in the TSHR gene found in the current case report also resulted in mild clinical features.
Genetic hyperthyroidism associated with activating mutations in the TSHR gene includes FNAH, PSNAH, and autonomous adenoma [12]. Although these diseases show similar pathophysiology, the level of activation of TSHR may vary depending on the type of mutation. The strength of TSHR in vitro is stronger in PSNAH than in FNAH, leading to more severe clinical symptoms and earlier onset [12,13].
Infants born to mothers with Graves' disease often show transient hyperthyroxinemia without any clinical signs, and they usually have a good prognosis [14]. Moreover, thyroid function test results return to normal between weeks 3 and 12, when maternal TSAbs that have crossed the placenta are lost [15]. In our study, the mother suspected personal hyperthyroidism secondary to Graves' disease, and the patient did not have clinical symptoms. Therefore, it was initially thought that the patient had hyperthyroidism because of the maternal TSAbs that had crossed the placenta and were lost as FT4 normalized, despite continued suppression of TSH. However, FT4 started increasing again after 4 months of age, and TSH remained suppressed, suggesting non-autoimmune hyperthyroidism. Nevertheless, diagnostic testing is not easy, and genetic analysis to identify an activating mutation in the TSHR gene is not performed routinely.
Thomas et al. [16] published the first report of FNAH in 1982; at the time, FNAH was diagnosed in cases where non-autoimmune hyperthyroidism was segregated within families. The first identification and report of a TSHR germline mutation were by Duprez et al. [17] in which p.Val509Ala and p.Cys672Tyr mutations were found in 2 French family pedigrees. The age of onset and clinical manifestations varied greatly within the same pedigree. Karges et al. [18] reported the same mutation with a novel mechanism of G protein-coupled receptor activation. The index patient with p.Val509Ala mutation showed hyperactivity and insomnia starting at 6 months of age and was diagnosed with congenital hyperthyroidism at 4 years of age. The paternal grandmother, who had an identical mutation, was diagnosed with follicular thyroid cancer at 60 years of age, demonstrating that persistent hyperthyroidism is a predisposing factor for thyroid neoplasia [18]. Therefore, early diagnosis and appropriate treatment are of utmost importance for improving outcomes in patients with non-autoimmune hyperthyroidism.
Treatment of non-autoimmune hyperthyroidism starts with antithyroid drugs (ATDs), including propylthiouracil and methimazole, and beta-blockers can also be used. However, because long-term treatment with ATDs may cause recurrence and goiter growth, it is not recommended [4,19]. Moreover, the risks of agranulocytosis and hepatitis should be considered as major side effects of ATD treatment [20]. Therefore, thyroidectomy is recommended as first-line treatment in patients with non-autoimmune hyperthyroidism, and radioiodine treatment can be used in patients 5 years and older [4,19,20]. Our patient is currently 2 years 10 months old, and he has been treated with methimazole and shown no side effects. However, the patient will require long-term follow-up to monitor the side effects of ATD treatment; consequentially, surgical treatment and radioiodine therapy should be considered.
In conclusion, we conducted WES using tissues obtained through buccal smears and confirmed FNAH caused by an activating TSHR germline mutation. Although 29 types of FNAH with activating TSHR gene mutations have been reported through 2018, no cases of FNAH associated with the A627V have been reported in other countries, and our case the second in South Korea. Based on the current case and previous report, the A627V variant in the TSHR gene is thought to cause mild clinical symptoms. However, more cases will be required to evaluate genotype-phenotype correlations. When patients with hyperthyroidism are negative for TSAbs and the cause is unclear, non-autoimmune hyperthyroidism caused by germline activating mutations of the TSHR gene should be considered because accurate diagnosis influences long-term treatment and prognosis of patients.
Go to :
 XML Download
XML Download