

 PDF
PDF Citation
Citation Print
Print



Introduction
Cytochrome P450 oxidoreductase (POR) deficiency (OMIM #613571) is a rare autosomal recessively-inherited form of congenital adrenal hyperplasia that was first reported in 1985 [1]. POR deficiency is caused by mutations in the POR gene encoding an electron donor for all microsomal P450 enzymes, including CYP21A2, CYP17A1, CYP51A1, and CYP19A1, as well as some non-P450 enzymes such as squalene epoxidase [2].
The disorder is characterized by clinical and endocrine features of both CYP17A1 (17α-hydroxylase/17,20-lyase) and CYP21A2 (21-hydroxylase) deficiencies, skeletal dysplasia, maternal virilization during pregnancy, ambiguous genitalia in female fetuses by virilization, and in male fetuses by undervirilization [3]. Combined deficiencies of the steroidogenic enzymes CYP17A1 and CYP21A2 result in glucocorticoid deficiency and impaired synthesis of sex steroids leading to ambiguous genitalia. CYP21A2 is required for synthesis of glucocorticoid and mineralocorticoid, whereas CYP17A1 is present in the adrenal glands and gonads catalyzing both 17α-hydroxylase and 17,20-lyase activities [4]. Underdeveloped male external genitalia in patients with 46, XY karyotypes can be explained by a deficiency of CYP17A1, resulting in decreased androgen production. The virilization of the female fetus may be explained by the synthesis of 5α-dihydrotestosterone through the "backdoor pathway" in the fetal gonads, which is active only during the fetal period [5]. CYP19A1 is an enzyme responsible for the conversion of androgens to estrogens and depends on electron transfer via POR. POR deficiency in the fetal placenta impairs the conversion of androgen to estrogen, resulting in maternal virilization during pregnancy [6].
Skeletal malformation is a distinctive clinical feature of POR deficiency, but the underlying mechanism remains elusive. One hypothesis is that the impaired function of CYP26B1, the PORdependent enzyme that degrades retinoic acid, produces an inappropriately high level of retinoic acid during embryogenesis. Retinoic acid can cause a wide range of limb malformations [7]. Squalene epoxidase and CYP51A1 are POR-dependent enzymes involved in cholesterol synthesis. In POR deficiency, impaired functions of squalene epoxidase and CYP51A1 cause defective cholesterol synthesis, resulting in defective signaling by the hedgehog protein, which is critical in the growth, patterning, and morphogenesis of many embryonic structures [8].
The prevalence of POR deficiency has yet to be determined. Since mutations in POR were identified in 2004 [9], nearly 140 cases of POR deficiency have been reported in the literature [10]. Our group reported a case of POR deficiency with skeletal phenotype in Korea [11]. In this study, we describe the clinical, biochemical, and molecular characteristics of four patients with POR deficiency in Korea.
Materials and methods
1. Subjects
This study included 4 unrelated patients (2 males and 2 females) with POR deficiency. Their clinical features, biochemical profiles, and radiologic findings were collected by retrospective chart review. Each diagnosis of POR deficiency was confirmed by molecular analysis of the POR gene. Standard deviation scores (SDS) for height and weight were calculated based on the Korean National Growth Charts for children and adolescents [12]. Subject 1 was previously reported [11] but was included in this study to delineate the clinical spectrum of POR deficiency in Korea. This study was approved by the Institutional Review Board at Asan Medical Center, Seoul, Korea. Blood samples for DNA analysis were collected after obtaining informed consent from the participants' parents.
2. Mutation analysis of the POR gene
Genomic DNA was extracted from peripheral blood leukocytes using the Gentra Puregene Blood Kit (QIAGEN, Hilden, Germany). Direct sequencing of the POR gene was performed using specific oligonucleotide primers. All 16 coding exons and the exon–intron boundaries were amplified by polymerase chain reaction (PCR) using primers designed with Primer3 cgi version 3.0 (http://bioinfo.ut.ee/primer3-0.4.0/). Amplified PCR products were directly sequenced using the BigDye Terminator v3.1 Cycle Sequencing Kit and an ABI3130x1 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA).
Results
1. Clinical and biochemical characteristics of patients with POR deficiency (Table 1)
Subject 1, a 7-month-old girl, was referred for evaluation of ambiguous genitalia and multiple anomalies. She was born at 40 weeks of gestation with a birth weight of 3.5 kg (0.57 SDS) from nonconsanguineous Korean parents. The patient's blood pressure was 98/68 mmHg, and height and weight were 69.8 cm (1.09 SDS) and 7.1 kg (-0.61 SDS), respectively. Head circumference was 47 cm (3.16 SDS). She showed brachycephaly, frontal bossing, proptosis, and midfacial hypoplasia with a depressed nasal bridge and low-set, dysplastic ears. Radiographs showed bilateral radiohumeral synostosis. The patient had clitoromegaly with partial labial fusion and a common urogenital sinus. Basal adrenocorticotropic hormone (ACTH) level was 290 pg/mL (normal, 10–60 pg/mL). A rapid ACTH stimulation test showed baseline and stimulated 17-hydroxyprogesterone (17-OHP) levels (normal, <6 ng/mL) of 22.4 ng/mL and 36.3 ng/mL, respectively, whereas the basal serum cortisol level of 9.3 μg/dL (normal, 2.8–23 μg/dL) was not increased after stimulation (9.6 μg/dL), suggesting adrenal insufficiency. Serum electrolytes were normal with 138 mmol/L of sodium and 5.1 mmol/L of potassium (Table 2). The karyotype was 46,XX. Glucocorticoid replacement with a physiologic dose of hydrocortisone (15 mg/m2/day) was started. She underwent genitoplasty and vaginoplasty at 17 months and rhinoplasty at 5 years of age. She was also diagnosed with vesicoureteral reflux at 7 months of age. She had recurrent otitis media in both ears, which eventually progressed to otosclerosis of the stapes and hearing loss at the age of 7. At her current age of 11 years, height and weight were 144.8 cm (-0.57 SDS) and 45.3 kg (0.52 SDS), respectively. She does not show intellectual disability, and her breast development is still Tanner stage I.
Subject 2, an 80-day-old girl, was referred to our institute due to ambiguous genitalia. She was born at 38 weeks of gestation from nonconsanguineous Korea parents and weighed 2.86 kg (-0.84 SDS) without any perinatal problems or maternal virilization. At the first visit, her height and weight were 60.5 cm (1.68 SDS) and 5.9 kg (1.08 SDS), respectively. The patient's blood pressure was 111/66 mmHg, which was above the 90th percentile for her age- and sex-matched reference [13]. Her external genitalia was partially virilized with clitoromegaly and partial fusion of the labia majora with a common urogenital sinus. No other skeletal anomalies or facial dysmorphism were found. Basal ACTH (415 pg/mL), plasma renin activity (PRA) (6.1 ng/mL/hr) (normal, normal 2.35-3.7ng/ml/hr), and 17-OHP (36.8 ng/mL) levels were elevated without electrolyte imbalance. Morning serum cortisol level was 15.4 μg/dL. Serum cortisol level increased from 13.4 μg/dL to 26.2 μg/dL after a rapid ACTH stimulation test. Chromosome analysis showed a normal female karyotype of 46,XX. The patient underwent vaginoplasty at 8 months of age. She had hypertension during the follow-up period with a peak blood pressure of 130/54 mmHg at 7 months of age. At her current age of 5 years, her height and weight are 118.4 cm (1.89 SDS) and 19.25 kg (0.14 SDS), respectively, with normal developmental milestones. She has been treated with oral hydrocortisone (6.25 mg/m2/day) and has normal blood pressure for her age.
Subject 3 was born at 3.11 kg (-0.49 SDS) in the 38th week of gestation without any perinatal problems from nonconsanguineous Korean parents. He had hypospadias and cryptorchidism without any skeletal abnormalities or facial dysmorphism. The neonatal screening test showed an elevated 17-OHP level of 17.6 ng/mL. Initial ACTH and cortisol levels were 90.6 pg/mL and 25.2 μg/dL, respectively. PRA (42.8 ng/mL/hr) levels were elevated. Serum electrolytes were normal. His karyotype was 46,XY. Glucocorticoid was started at 3 months of age at a dose of 14.9 mg/m2/day. He underwent multiple surgeries to repair hypospadias and urethral fistula until 35 months of age. His blood pressure was normal during the follow-up period. At his current age of 39 months, his height and weight are 104.9 cm (1.61 SDS) and 21.0 kg (3.09 SDS), respectively. He has been treated with hydrocortisone 3.3 mg 3 times a day (12.8 mg/m2/day) without any adrenal crisis events.
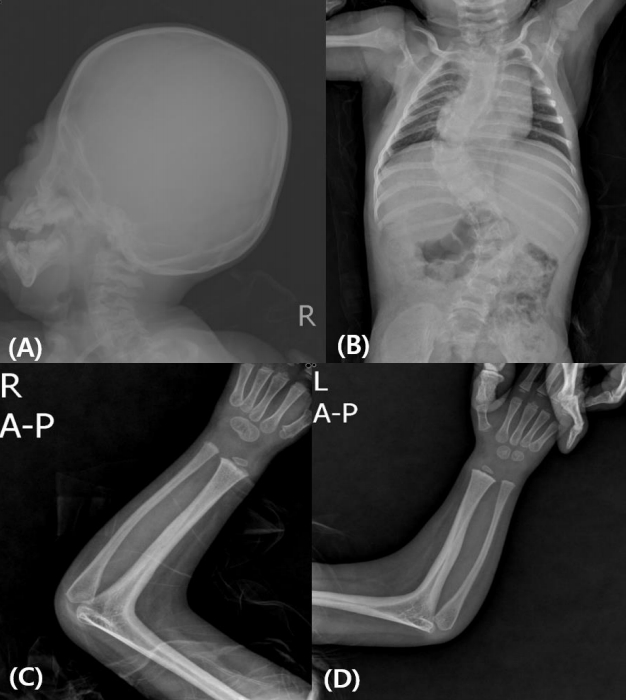
Subject 4 was born at 39 weeks of gestation with a birth weight of 2.64 kg (-1.56 SDS) from nonconsanguineous Korean parents. He showed underdeveloped male genitalia with hypospadias and micropenis. Dysmorphic face with brachycephaly and midfacial hypoplasia was prominent. Radiologic findings showed radiohumeral synostosis with contracture of both elbow joints, early fusion of cranial sutures, and congenital scoliosis (Fig. 1). Echocardiography revealed atrial septal defect (3.4 mm) and ventricular septal defect of approximately 5 mm. He underwent cardiac repair surgery at 1 month of age. Plasma ACTH (940.4 pg/mL) and serum 17-OHP (1,050 ng/mL) levels were elevated at initial presentation. PRA (22 ng/mL/hr) was elevated, but serum electrolytes were normal. He had a normal male karyotype of 46,XY. Hearing aids were applied at 9 months of age due to sensorineural hearing loss in both ears. His blood pressure was not measured regularly, but at 28 months of age, his blood pressure was 115/69 mmHg, which was above the 95th percentile for his age-, sex-, and heightmatched reference [14]. At present, he is 31 months old and has been treated with physiologic doses of hydrocortisone (15 mg/m2/day) and has a height of 86.8 cm (SDS -1.69) and weight of 10.2 kg (SDS -2.37). He shows delayed language development, making only inappropriate sounds at 31 months old.
2. Molecular analysis of the POR gene (Table 3)
Direct sequencing of POR identif ied two dif ferent mutations from 4 unrelated patients, c.1370G>A (p.R457H) and c.1329_1330insC (p.I444Hfs*6) (Table 3). Both were previously reported as pathogenic mutations [15-17]. Subjects 1 and 4 were compound heterozygotes for c.1370G>A (p.R457H) and c.1329_1330insC (p.I444Hfs*6). Subjects 2 and 3 were homozygotes for c.1370G>A (p.R457H). POR c.1370G>A (p.R457H). The p.R457H mutation in POR, known as a global founder mutation [15], was the most common mutation in studied patients, with 75% allele frequency (6/8 alleles).
Discussion
In this study, all patients with POR deficiency manifested typical clinical features, such as adrenal insufficiency and ambiguous genitalia regardless of sex or skeletal phenotypes. Genetic males (46,XY) had underdeveloped male genitalia with micropenis and hypospadias, while 2 genetic females (46,XX) were virilized with clitoromegaly and urogenital sinus. Interestingly, virilization of the female infants did not progress postnatally, which is a distinctive feature of POR deficiency compared with other forms of congenital adrenal hyperplasia such as 21-hydroxylase deficiency. Maternal virilization during pregnancy was not documented in our patients.
Female patients are virilized at birth due to the "backdoor pathway" in fetal gonads to produce androgen, bypassing the conventional pathway [18]. During the first several months of life, gonadal expression of the 5α-reductase type 1 and 3α-hydroxysteroid dehydrogenase are replaced by the 5α-reductase type 2, which no longer uses 17-OHP as a substrate to produce 5α-dihydrotestosterone. As a result, the backdoor pathway of androgen synthesis ceases [4]. Therefore, unlike girls with untreated 21-hydroxylase deficiency, patients with POR deficiency are no longer virilized after birth.
POR deficiency is also associated with delayed puberty in both sexes. Delayed puberty is caused by insufficient sex hormone production in the gonads resulting from the defective functions of CYP19A1 and/or CYP17A1 [19]. Subject 1, an 11-year-old girl, did not develop signs of pubertal onset, necessitating estrogen replacement therapy. The other patients are too young for their pubertal development to be evaluated, and should be closely monitored to observe whether they exhibit normal sexual development.
The diagnoses of the 4 patients in this study were confirmed by the molecular analysis of POR. The patients with milder clinical phenotypes were homozygous for p.R457H, while the other 2 patients (subjects 1 and 4) were compound heterozygotes of p.R457H and p.I444Hfs*6, and displayed more severe phenotypes including skeletal abnormalities. According to a previous study of genotype-phenotype correlations in Japanese patients [17], homozygotes with the p.R457H mutation are predicted to have greater enzymatic activity of POR. Clinical symptoms such as skeletal anomaly, adrenal dysfunction, and delayed pubertal development are more severe in patients with frameshift mutations than in p.R457H homozygotes [20]. Similarly, subjects 2 and 3 had homozygous mutations of p.R457H and displayed milder clinical phenotypes. Subjects 1 and 4 had compound heterozygous mutations with frameshifts and exhibited other congenital anomalies involving heart, kidney, and hearing. Conductive hearing loss and vesicoureteral reflux are often present in patients with these mutations, according to the literature [21,22].
Patients with POR deficiency have a combined deficiency of CYP21A2 and CYP17A1, leading to impaired glucocorticoid production in the adrenal glands. Adrenal involvement usually presents with partial dysfunction [20]. A life-threatening adrenal crisis is not a major clinical manifestation of POR deficiency, but there is a possibility of adrenal crises during stressful conditions such as febrile illnesses or major surgery [23]. The degree of 17-OHP elevation in newborn screening tests tends to be less remarkable than in patients with classic 21-hydroxylase deficiency [20]. A daily supplement of hydrocortisone is not always essential for all patients with POR deficiency. Steroid replacement should be tailored individually according to clinical features and hormonal findings. Mineralocorticoid dysfunction is not apparent in POR deficiency. However, the p.A287P mutation, the most frequent mutation in patients of western Eurasian descent [23], is associated with increased blood pressure by increased deoxycorticosterone [24]. Two of our patients were observed to have high blood pressure during follow-up. Thus, it is necessary to monitor blood pressure regularly in POR deficiency patients.
The diagnosis of POR deficiency is not difficult in patients with Antley-Bixler syndrome phenotypes. However, milder forms of POR deficiency that manifest with slightly elevated 17-OHP and ambiguous genitalia at birth can be misdiagnosed with other types of congenital adrenal hyperplasia. Moreover, considering the wide phenotypic spectrum of POR deficiency, mildly affected patients presenting with only primar y amenorrhea or partial glucocorticoid deficiency could remain undiagnosed [21]. Therefore, molecular analysis of the POR gene is crucial for making a correct diagnosis.
Two other cases with POR deficiency were previously reported in Korea [25,26]. One newborn boy presented with respiratory difficulty after birth due to glossoptosis and choanal atresia [25]. He manifested adrenal insufficiency with skeletal anomalies such as early fusion of the left coronal suture and bilateral radio-humero-ulnar synostosis. He was a compound heterozygote for p.R457H and p.Q555Pfs*19 in POR, inherited from his father and mother, respectively. The other patient was a 21-year-old woman with delayed puberty, loss of pubic hair, and skeletal anomalies [26]. Her initial serum 17-OHP was 5.31 ng/mL. Serum cortisol was not elevated following ACTH stimulation. Pelvic ultrasonography revealed an underdeveloped uterus. A compound heterozygous mutation of c.529G>C (p.G177R) and c.1370G>A (p.R457H) in POR was identified. Including the subjects in the present study, all Korean patients harbored p.R457H, supporting the hypothesis that p.R457H is the global founder mutation (Table 3).
The main limitation of this study is the retrospective observation of a small number of patients, which makes it difficult to comprehensively describe the variable phenotypes associated with POR deficiency. The follow-up periods were also variable, from 1 year to 11 years, and therefore not adequate to delineate long-term clinical outcomes of POR deficiency. Therefore, long-term observational studies with larger numbers of patients are needed to clarify the clinical course, including growth and pubertal development, as well as effects on fertility associated with this rare form of congenital adrenal hyperplasia.
In conclusion, we report 4 patients with POR deficiency confirmed by mutation analysis of the POR gene. All 4 patients presented with ambiguous genitalia and adrenal insufficiency requiring daily supplementation of hydrocortisone. Two showed skeletal abnormalities consistent with Antley-Bixler syndrome. The phenotypic spectrum of POR deficiency is broad, ranging from adrenal insufficiency and ambiguous genitalia to severe skeletal malformations. Clinical and endocrine features are similar to those of 21-hydroxylase or 17α-hydroxylase deficiency. Therefore, molecular analysis of the POR gene is critical for the confirmatory diagnosis of POR deficiency.
 XML Download
XML Download