

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Resistance to thyroid hormone (RTH) is a rare inherited syndrome characterized by diminished response of target tissue to thyroid hormone. The patients with RTH have elevated serum levels of free thyroxine (fT4) and triiodothyronine (T3) in the absence of thyroidstimulating hormone (TSH) suppression1). The prevalence of RTH is approximately 1:40,000 births2). Its incidence is equal in males and females. The inheritance is autosomal dominant. Approximately 85% of the cases have a mutation in the thyroid hormone receptor beta (THRβ) gene, and more than 100 mutations have been discovered. The clinical manifestations vary. Goiter is the most common clinical finding. Other symptoms and signs include sinus tachycardia, learning disabilities, developmental delay, and delayed bone age3).
Here, we report the case of a child, who presented with goiter, with RTH due to a novel mutation in the THRβ gene.
Go to : 
Case report
A 9-year-old girl was referred to Chosun University Hospital because of abnormal thyroid function test results. She noted an asymptomatic goiter 7 days ago. The results of her thyroid function tests revealed that the serum levels of fT4 and TSH were 7.12 ng/dL and 12.4 uIU/mL, respectively.
She was born at full term and had no remarkable medical history of hyperactivity or learning problem. She had average performance at school. There was no family history of thyroid disorders. Her physical examination revealed the following: height, 141 cm (75th-90th percentiles); weight, 37 kg (75th-90th percentiles); pulse rate, 102 beats/min (average rate, 70-110 beats/min); blood pressure, 110/70 mmHg; and body temperature, 37.3℃. The thyroid gland was diffusely enlarged. There was no trill or exophthalmos.
The complete blood count and blood chemistry examination showed normal results. Her thyroid function test results revealed that the serum levels of fT4, T3, and THS were 9.89 ng/dL (reference range, 0.7-1.8 ng/dL), 403.2 ng/dL (reference range, 60-190 ng/dL), and 11.82 µIU/mL (reference range, 0.25-4.0 µIU/mL), respectively. She tested negative for antithyroglobulin antibody, antimicrosome antibody, and TSH receptor antibody. Her serum levels of prolactin, luteinizing hormone, and folliclestimulating hormone were within the reference range. She underwent a thyrotropin-releasing hormone (TRH) stimulation test. After receiving 200 µg/m2 of TRH, the TSH showed a normal response (TSH at baseline, 8.38 µIU/mL; TSH at 15 minutes, 17.36 µIU/mL; TSH at 30 minutes, 21.70 µIU/mL; TSH at 60 minutes, 19.42 µIU/mL; TSH at 90 minutes, 17.89 µIU/mL; TSH at 120 minutes, 4.95 µIU/mL).
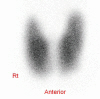
The 99m Technetium thyroid scan revealed diffuse enlargement with increased uptake in both lobes of thyroid gland; no cold nodules were observed (Fig. 1). A magnetic resonance imaging scan showed no abnormalities of the pituitary.
A polymerase chain reaction sequencing of the THRβ gene of the patient showed a novel mutation in exon 9. This was a heterozygous C-to-T transition mutation in the 327th codon, substituting threonine for isoleucine (c.980C>T; p.Thr327Ile) (Fig. 2). Analysis with the software program, Polyphen and SIFT indicated that this sequence change is probably damaging. Her parents were also examined for this mutation; the same mutation was found in her father, who was asymptomatic. The patient remained asymptomatic and did not need any treatment at the 12-month follow-up.

Go to :
Discussion
RTH is an inherited syndrome, first described by Refetoff et al.4) in 1967, characterized by refractoriness of target tissues to thyroid hormone.
Thyroid hormone receptors (TRs) are ligand-dependent transcription factors that mediate the activity of T3. TRs are encoded by 2 genes, designated as α and β genes. Four different TRs have been identified: α-1, α-2, β-1 and β-23). Each TR isoform has tissue specific expression that varies with the stage of development. Thyroid hormone receptor alpha-1 (THRα1) is highly expressed in the heart, bone, and brain; THRβ1 is more abundant in the liver, kidney, and thyroid; and THRβ2 expression is limited to the pituitary, hypothalamus, and ear5). About 90% of RTH patients have mutations in the THRβ gene, and at least 128 mutations in this gene have been reported thus far3). RTH without mutations occurs in approximately 10% of the cases. Recently, mutations in the THRα gene have been identified in some case reports6,7). These patients show different phenotypes from the RTH patients with THRβ gene mutation, with relatively low serum T4 and high serum T3 levels, growth and developmental retardation, delayed bone development, and constipation.
Including our case, 11 cases of RTH have been reported in the literature in Korea8); 8 patients had known missense mutations in the THRβ gene, 2 patients had novel mutations_insertion and missense mutation (our case) in the THRβ gene9) and 1 patient had no mutation10).
Familial occurrence of RTH has been documented in approximately 75% of cases, and approximately 20% of the patients are de novo cases3).
The hallmark of RTH is the paucity of clinical features of thyroid dysfunction despite increased serum T4 and T3 levels. When present, the clinical feature is highly heterogeneous owing to variable degrees of peripheral resistance in different patients, as well as variable resistance in different tissues within a single individual11). The clinical features are goiter, tachycardia, hyperactivity, developmental delay, and learning disability. The diagnosis of RTH is based on clinical findings, standard laboratory tests, and genetic studies. The finding of elevated serum thyroid hormone levels association with unsuppressed TSH usually leads to the diagnosis3).
It is important to accurately identify RTH, as misdiagnosis could lead to unnecessary treatments such as thyroidectomy or radioiodine. Amor et al.12) reported that 19% of the patients had undergone some type of thyroidal ablative therapy before diagnosis, similar to a result published 15-20 years earlier. Treatment is not required in most patients because the elevated thyroid hormones levels compensate for the partial tissue resistance. Beta-adrenergic blockers relieve sinus tachycardia. Supraphysiological doses of liothyronine, given as a single dose every other day, are effective in reducing goiter without causing side effects13). In children, particular attention must be paid to growth, bone maturation and mental development3).
In conclusion, we report a novel mutation in the THRβ gene in a 9-year-old girl. RTH is very rare and must be considered in patients who present with goiter and increased thyroid hormone levels with unsuppressed TSH.
Go to :
 XML Download
XML Download