

 PDF
PDF Citation
Citation Print
Print



Introduction
Diabetic foot ulcers (DFUs) are ulcers that arise on the feet of individuals with diabetes and are a major concern. These ulcers stem from the deterioration of the skin or mucosal tissue on the feet and are particularly susceptible to exacerbation by conditions such as diabetic neuropathy and peripheral vascular disease. Upon occurrence, DFUs can culminate in foot amputation, inflicting a substantial psychological burden on patients. This unfortunate outcome can lead to a reduction in one's daily activities, culminating in a decline in both physical capabilities and social engagement, leading to a serious reduction in the patient's quality of life and high financial costs for society [1].
As the average age of the population increases worldwide, the number of patients with diabetes, and accordingly, the number of patients with DFUs, is also increasing. The lifetime prevalence of DFUs in the population with diabetes is 15% to 25%, and the recurrence rate of DFUs within 5 years is high, ranging from 50% to 70% [2-4].
Understanding the pathophysiology of DFUs is crucial for effective management and prevention, facilitating early diagnosis, informed decision-making, personalized treatment, improved wound healing, fewer complications, preventive actions, and research advancements.
Pathophysiology of diabetic foot
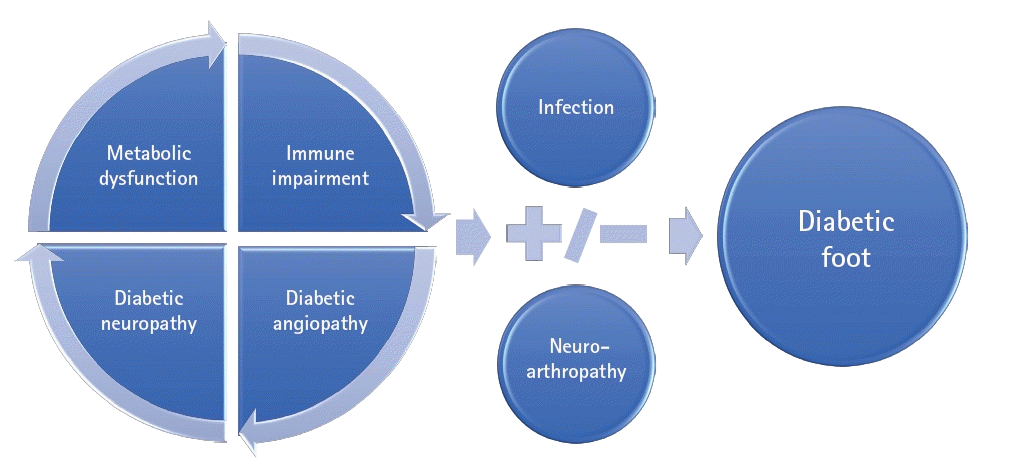
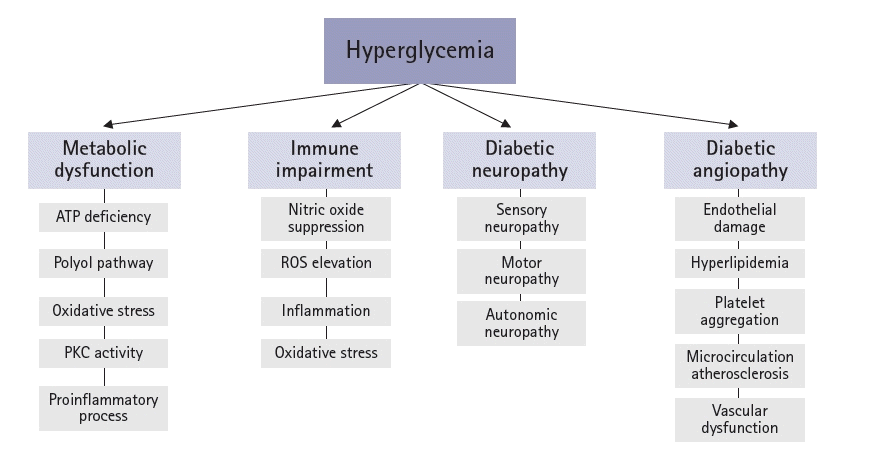
The pathophysiology of diabetic ulcers involves metabolic causes, neuropathy, angiopathy, and changes in the immune system. The interaction between metabolic dysfunction, diabetic immunopathy, diabetic neuropathy, and diabetic angiopathy promotes the development and progression of diabetic foot infections (DFIs) and may lead to diabetic neuroarthropathy (Figs. 1, 2).
1. Metabolic dysfunction
Diabetes affects epineural microvessels and reduces blood supply to the nerves of patients with diabetes [5]. Apart from the vascular supply, the peculiar anatomy of the peripheral nervous system may explain the predisposition of its most distal parts to diabetes. The axon is covered with Schwann cells, but the distal axon is too weak because the neuronal cell body is relatively small compared to the very long axon neurite. Therefore, distal axons are vulnerable when diabetes affects nerves [6].
The processes by which hyperglycemia causes peripheral nerve damage are related to adenosine triphosphate (ATP) deficiency, the polyol pathway, oxidative stress, protein kinase C (PKC) activity, and proinflammatory processes.
An insufficient ATP supply hampers axonal transport, particularly in mitochondria-rich axons that provide nerve energy, thus promoting axonal injury and diabetic neuropathy. The inability to counter excessive oxidative stress due to inadequate ATP levels damages the axons during hyperglycemia, causing axonal degeneration or apoptosis [7]. Oxidative stress negatively affects multiple biochemical pathways.
The polyol pathway involves the conversion of glucose to sorbitol by aldose reductase (AR) and the subsequent conversion of sorbitol to fructose by sorbitol dehydrogenase. In diabetes, elevated glucose levels boost the affinity of AR for glucose, leading to increased sorbitol production. Accumulated sorbitol reduces Na+K+-ATPase activity, thereby diminishing nerve cell reserves and conduction velocities. The hyperglycemia-induced polyol pathway also increases oxidative stress due to nicotinamide adenine dinucleotide phosphate (NADPH) depletion via the pentose phosphate pathway, which is essential for glutathione generation [8]. Excess fructose accelerates glycation and NADPH consumption and exacerbates intracellular oxidative stress [9]. This disturbance results in decreased antioxidant levels and increased production of reactive oxygen species (ROS), which play pivotal roles in diabetes-related complications. The activation of AR increases polyol flux and causes neuropathy; however, neuropathic changes have also been observed in AR-deficient diabetic mice [10]. Further research is required to establish a direct link between AR and diabetic neuropathy.
PKC is a member of the serine/threonine protein kinase family [11] and is involved in various cellular responses associated with diabetes [12]. Hyperglycemia triggers glycolysis and activates PKC [13]. Vascular dysfunction caused by PKC activation promotes diabetic microvascular complications, which primarily alter blood flow [14], extracellular matrix synthesis, basement membrane thickening [15], vascular permeability [16], and angiogenesis [17].
Low-grade intraneural inflammation is an aspect of diabetic neuropathy. Systemic proinflammatory activity in human sensorimotor diabetic neuropathy has recently been reported [18]. This mild inflammatory process is a common terminal pathway in diabetic neuropathy and is associated with the degeneration of intraepidermal nerve fibers.
2. Diabetic immunopathy
In the context of hyperglycemia, the suppression of endothelial nitric oxide (NO) production inhibits NO synthase, leading to an elevated level of ROS, notably superoxide radicals. This heightened ROS level subsequently triggers an increase in hydrogen peroxide concentrations. Consequently, highly reactive hydroxyl radicals can be formed that cause cellular damage. The combined action of NO and superoxide results in the production of peroxynitrite, which, in turn, affects endothelial vasodilation and mediates lipid peroxidation. This process sets the stage for heightened concentrations of low-density lipoproteins, followed by the development of microcirculation atherosclerosis, elevated inflammation, abnormal intimal growth, platelet aggregation, and thrombosis [19].
Hyperglycemia contributes to excess hydrogen peroxide, intensifying oxidative stress and related products [20]. These products stimulate the generation of advanced glycation end-products (AGEs) [21]. Decoupling of NO synthase reduces NO production, resulting in impaired wound healing [22]. In the context of wound healing, the controlled generation and elimination of ROS are vital. However, diabetic wounds exhibit elevated ROS levels, which further impede the healing process. The heightened presence of ROS not only slows wound healing but also leads to excessive oxidative stress.
3. Diabetic neuropathy
Peripheral neuropathy is the most common intractable complication of diabetes [23,24]. More than 60% of DFUs result from an underlying neuropathy [25]. The duration of diabetes and glycated hemoglobin levels are strongly associated with the prevalence of neuropathy [26,27].
Blood supply to the peripheral nerves is insufficient, blood flow is easily damaged, and automatic regulation of blood flow is impaired. These anatomical features enable us to understand why peripheral nerve neuropathy differs from other complications [28]. These features make peripheral nerves vulnerable to ischemia.
In diabetic neuropathy, damaged nerve endings lead to pain perception due to disrupted action potentials, inducing hyperexcitability [29]. Upregulated voltage-gated sodium channels (Nav), including Nav1.3 and Nav1.7, in diabetic animals influence diabetic neuropathy progression, while reduced Nav1.8 and Nav1.6, along with abnormal phosphorylation of Nav1.6, Nav1.7, and Nav1.8, contribute to nociceptive nerve fiber irregularities [30,31]. Patients with diabetic neuropathy display increased nodal Na+ currents that intensify peripheral nerve hyperexcitability.
Altered K+ voltage-dependent channels (Kv) affect resting membrane potential and increase neuronal excitability [32]. Reduced Kv currents in diabetic rats elevate excitability and peptide release, and these factors could contribute to peripheral nerve hyperexcitability in diabetes.
The development of neuropathy in affected patients results from hyperglycemia-induced metabolic abnormalities [33,34]. Diabetic neuropathy affects the sensory, motor, and autonomic nervous systems.
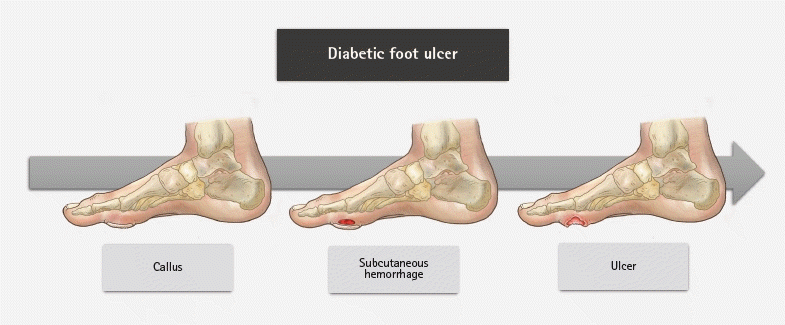
The evidence of sensory neuropathy includes hyperalgesia, paresthesia, and allodynia. Sensory neuropathy causes loss of protective sensations. As a result, the risk for trauma is significantly elevated [35,36] and injuries often go unnoticed for weeks. Motor neuropathy can manifest as atrophy of the small foot muscles, resulting in malpositioning of the toes (claw toe) [37]. These muscle changes can cause foot deformities leading to biomechanical abnormalities. Glycosylation of tendons induces stiffness and shortening, potentially giving rise to foot deformities, such as claw toes and hammer toes, along with Achilles tendon stiffening, which elevates the pressure on the forefoot [38]. The combination of sensory and motor peripheral neuropathy results in an unequal foot load with an unsteady gait. Over time, hyperkeratosis develops because of neuropathy and an elevated plantar pressure load. Autonomic neuropathy reduces the function of sweat and sebaceous glands of the feet, resulting in dry skin and fissures. Furthermore, it diminishes the neuroinflammatory responses to noxious stimuli [39]. Consequently, the natural ability of the foot to moisturize is lost, and the skin becomes more susceptible to injury and infection. The interaction between sensory, motor, and autonomic neuropathy can form a callus in the foot. After repeated exposure to external or minor trauma, skin ulcers form through subcutaneous hemorrhage (Fig. 3) [40].
4. Diabetic angiopathy
Hyperglycemia causes endothelial damage, dyslipidemia, and increased platelet viscosity and activity, leading to atherosclerosis. Hyperglycemia also causes peripheral vascular changes, resulting in endothelial cell dysfunction and decreased vasodilator secretion [25,41]. Peripheral vascular constriction and hypercoagulation lead to ischemia, which increases the risk of skin ulcers. Peripheral arterial disease is an important cause of foot ulcers in approximately 50% of cases [42]. An insufficient peripheral blood supply delays wound healing and exacerbates infections [43].
In diabetic neuropathy, the perception of pain arises from impaired nerve endings that disrupt the normal progression of action potentials. Atherosclerosis is the primary contributor to vascular mortality in type 2 diabetes. This process is driven by endothelial cell dysfunction, a consequence of factors like hypertension, insulin resistance, and hyperglycemia. The resulting dysfunction not only hampers the production of NO by endothelial cells but also triggers signals that encourage the proliferation of vascular smooth muscle cells through the mitogen-activated protein kinase pathway. Elevated glucose levels counteract the vasodilatory effects of NO and increase the levels of antifibrinolytic and prothrombotic molecules [4].
The intricate relationship among insulin resistance, hyperglycemia, and dyslipidemia underscores the nature of diabetes. Through extensive research, four interconnected pathways have been revealed, shedding light on the connection between hyperglycemia and endothelial cell damage, resulting in shared biochemical abnormalities. Elevated glucose exposure spurs the production of mitochondrial superoxide, thereby influencing downstream pathways including the hexosamine and PKC pathways. Hyperglycemia-induced mitochondrial generation of ROS affects both endothelial cells and platelets, inciting platelet aggregation and cytokine release [4,44].
In the context of diabetes, targeting the excessive production of mitochondrial ROS is promising for addressing vascular dysfunction. Gliclazide, a sulfonylurea, possesses antioxidant and anti-aggregation properties that can potentially enhance vascular regulation and mitigate oxidative stress. It effectively interferes with AGE-induced vascular endothelial growth factor expression while modulating signaling pathways intricately linked to diabetic angiopathy [45].
5. Diabetic foot infections
The interplay of metabolic factors, immunopathy, diabetic neuropathy, and diabetic angiopathy promotes the development and progression of infections, ischemic ulcers, and gangrene in diabetic individuals, potentially culminating in amputation (Fig. 4) [46,47].
A range of microorganisms can trigger DFIs (Fig. 3) [40], with Staphylococcus aureus being the most prevalent. Among DFIs, methicillin-resistant S. aureus (MRSA) is found in 16.78% to 30% of cases [48]. Although MRSA infections do not appear to affect mortality, they are associated with increased hospitalization rates and increased risks of limb amputation [49]. Amputation, as a preventive measure against the spread of infection, was shown to extend life expectancy by 2 years in half of the studied subjects with diabetes [50,51]. Nevertheless, the survival rate of patients with diabetes and ulcerative infections remains at only 56% 5 years after the initial onset of ulcers [51]. Collectively, these findings underscore the urgency of enhanced ulcer prevention and prompt diagnosis of DFIs.
6. Neuroarthropathy
Charcot neuroarthropathy, commonly referred to as Charcot foot, is a chronic and progressive degenerative arthropathy that arises from disrupted sensory innervation of the affected joint. This insidious and destructive condition primarily affects the foot bones, leading to deformities that can trigger ulcer formation and subsequent disabilities. Charcot foot development is characterized by joint subluxation and dislocation, bone osteolysis, fragmentation, and soft-tissue edema [47].
Deterioration of the autonomic nervous system in individuals with diabetes mellitus leads to increased local blood supply, resulting in elevated resting blood flow compared to that in individuals who are non-diabetic. This heightened blood flow combined with increased calcium dissolution promotes osteoclastic bone activity and damage. Another theory suggests that repeated minor trauma to the insensate joints contributes to fracture and disintegration.
In patients with Charcot foot, proinflammatory proteins exhibit distinct expression patterns under modulated stimulation. The production of proinflammatory cytokines, such as tumor necrosis factor-α and interleukin (IL)-1β, triggers uncontrolled osteolysis. These cytokines upregulate the expression of receptor activator of nuclear factor (NF)-κB ligand (RANKL), which in turn promotes osteoclast maturation through NF-κB activation, while the anti-inflammatory peptides IL-4 and IL-10 are downregulated.
A hallmark deformity associated with Charcot foot is midfoot collapse, often termed the “rocker-bottom” foot deformity. Hallux valgus and loose bodies in the joint cavity may also be present. These deformities increase susceptibility to recurrent ulcerations [52].
Conclusion
Hyperglycemia induces diabetic ulcers through several processes. The interactions among metabolic dysfunction, diabetic immunopathy, diabetic neuropathy, and diabetic angiopathy promote the development and progression of DFIs and may lead to diabetic neuroarthropathy. A direct and detailed approach to understanding the pathogenesis of DFUs will enable the development of new treatment methods and prevent the worst outcomes of diabetic ulcers such as major amputation.
 XML Download
XML Download