

 PDF
PDF Citation
Citation Print
Print



Introduction
Mesenchymal stem cells (MSCs), also known as mesenchymal stromal cells, are fibroblast-like multipotent adult stem cells with the capacity to self-renew. In recent decades, MSCs has gained significant attention due to their potential to regenerate damaged tissues by secreting paracrine factors (1). The therapeutic effects of MSCs are driven by complex mechanisms, such as their ability to differentiate into specific tissues, release active substances that contribute to their efficacy, and modulate an immune system (2-4). The investigation of the pharmacodynamics (PD) aspect of MSCs is challenging due to the intricate nature of these cells. However, preclinical pharmacokinetics (PK) studies, which aid in optimizing the necessary dosage regimen to attain therapeutic effects and determine cell distribution post-transplantation, are feasible.
Several delivery strategies have been employed to deliver MSCs to the central nervous system (CNS) (5-7). Intravenous injection is the least invasive method, but it is also the least efficient (8, 9) because the cells are largely trapped in the lung where they undergo cell death shortly (10, 11). More direct routes such as intra-cerebral, intra-ventricular, and intra-thecal injections have been tested to deliver therapeutic agents into the CNS (12, 13). However, the PK aspects of MSCs following intra-cranial transplantation remain poorly understood.
In this study, we systematically conducted a preclinical study to assess the biodistribution of MSCs that were locally transplanted into the brain, an immune privileged organ, in animal models with compromised immune res-ponses. We also employed a regimen involving the use of cytosine deaminase (CD) and 5-fluorocytosine (5-FC) to eliminate resident immune cells at the injection site. The CD enzyme converts non-toxic 5-FC into a cytotoxic anti-cancer drug, 5-fluorouracil (5-FU). MSCs expressing CD (MSCs/CD) can induce cell death in the neighboring bystander cells while simultaneously undergoing self-induced cell death (14-16). We examined the impact of 5-FC on local immune cells after local transplantation into the brain as well as the biodistribution of MSCs/CD.
The findings from our PK study focusing on the biodistribution of MSCs following transplantation into the brain will contribute to advancement of stem cell-based therapies for the treatment of neurological diseases in cli-nical applications.
Go to : 
Materials and Methods
Animals
All experimental procedures using animals were approved by the Institutional Animal Care and Use Committee of Ajou University School of Medicine, Korea (No. 2020-0009). Equal numbers of male and female nude mice, aged 8 weeks old (Hsd: Athymic Nude-Foxn1nu; Envigo) were used for stereotactic administration of MSCs or MSCs/CD cells in brain. The mice were housed with ad libitum access to food and water and maintained in a 12:12 hours light-dark cycle until being euthanized.
Human MSCs and MSCs/CD cells
Human MSCs were derived from the iliac crest’s bone marrow of a 19-year-old healthy donor as described previously (17) with approval from the Institutional Review Board of Ajou University Medical Center (No. AJIRB-BMR-KSP-20-040) with the informed consent of the patient. Briefly, mononucleate cells were maintained as adherent cultures in Dulbecco’s Modified Eagle’s Medium (Cat. No. LM 001-05; Welgene) supplemented with 10% fetal bovine serum (FBS, Cat. No. 16000-044; Gibco), 100 U/ml penicillin, 100 μg/ml streptomycin (Cat. No. 15140-122; Gibco) and 10 ng/ml basic fibroblast growth factor (Cat. No. 100-18B; PeproTech). MSCs/CD cells were prepared by transducing MSCs with retroviral vector encoding a bacterial CD gene as described previously (14).
Characteristics of MSCs and MSCs/CD
Mesodermal differentiation was carried out as described previously (18). Briefly, for adipogenic and osteogenic differentiations, naïve MSCs and MSCs/CD cells were plated at a density of 5×104 in a 24-well plate and allowed to grow to confluence. Then, the media was replaced every 2∼3 days with StemProTM osteogenesis differentiation media (Cat. No. A1007201; Thermo Fisher Scientific) and StemProTM adipogenesis differentiation media (Cat. No. A1007001; Thermo Fisher Scientific), respectively. After 14 days, the differentiated cells were washed with phosphate buffered saline (PBS) and then fixed with 10% neutral buffered formalin (Cat. No. 015MIRA01; BBC Bioche-mical). For adipogenesis, Oil Red O staining was performed. For osteogenesis, alizarin red S staining was performed. For chondrogenic differentiation, 3×105 cells were washed with PBS and centrifuged at 500 ×g for 5 minutes. The cell pellet was induced to undergo chondrogenic differentiation in StemProTM chondrogenesis differentiation media (Cat. No. A1007101; Thermo Fisher Scientific) for 4 weeks, with media replacement every 2∼3 days. After completion of chondrogenic induction, the pellet was washed with PBS, fixed in 10% neutral buffered formalin for 30 minutes at room temperature, washed again, and embedded in paraffin. The chondrogenic pellet was sectioned at a thickness of 5 μm, and the mid-section was deparaffinized and stained with Alcian blue solution for 1 hour at room temperature. After counterstaining with nuclear red for 3 minutes at room temperature, the sample was mou-nted using Shandon Synthetic MountantTM (Cat. No. 6769007; Thermo Fisher Scientific). The images of Oil Red O positive adipocytes, alizarin red positive osteocyte and Alcian blue positive chondrocyte were taken using an EVOS M5000 imaging system (Thermo Fisher Scientific).
To measure the expression of surface antigens in MSCs and MSCs/CD, fluorescence-activated cell sorting (FACS) analysis was performed as described previously (19). Brie-fly, cells were stained for 15 minutes at 25℃ with fluorochrome-conjugated antibodies against CD29 (Cat. No. 303003; BioLegend), CD90 (Cat. No. 559869; BD Bioscie-nces), CD105 (Cat. No. 323205; BioLegend), CD34 (Cat. No. 343505; BioLegend), CD45 (Cat. No. 304011; BioLegend), HLA-DR (Cat. No. 560896; BD PharmingenTM), and the isotype control. The cells were washed with PBS and suspended in flow cytometry staining buffer. Cells were analyzed using an Attune NxT Acoustic Focusing Cytometer (Thermo Fisher Scientific) with AttuneTM NxT software.
Quantitative polymerase chain reaction
The qPCR (quantitative polymerase chain reaction) was performed on the StepOnePlus Real-Time PCR System (Applied Biosystem) using 100 ng of genomic DNA (gDNA) in a 20 μl reaction mixture containing 10 μl of PowerSYBRⓇ Green PCR Master Mix (Cat. No.4367659; Applied Biosystem) and 0.5 μM each of the forward and reverse primer. The human Alu specific primers were 5’-CACCTGTAATCCCAGCACTTT-3’ (forward) and 5’-CCC AGGCTGGAGTGCAGT-3’ (reverse). The PCR protocol consisted of 10 minutes of denaturation at 95℃ followed 40 cycles of 95℃ for 15 seconds, 65℃ for 30 seconds, and 72℃ for 30 seconds. A standard curve was generated by diluting refence samples of human gDNA from MSCs in mouse gDNA, spanning a range of 0.01∼100 ng. To maintain a consistent total amount of DNA (100 ng/reaction), human gDNA samples were mixed with mouse gDNA.
Assessment of in vivo distribution of transplanted cells
MSCs or MSCs/CD were harvested, washed twice, and then resuspended in PBS at a density of (0.5×105 cells/μl PBS). The 6 μl of cell suspension was injected into striatum (anteroposterior, +0.05 cm; mediolateral, −0.18 cm; dorsoventral, −0.3 cm) of nude mouse using stereotaxic device (Stoelting Co.) at a rate of 0.3 μl/min. On day 0∼28 after the transplantation, gDNA was prepared from the ipsilateral and contralateral cerebral hemisphere, lung, and liver of the animals using the ReliaPrepTM gDNA Tissue Miniprep System (Cat no. A2051; Promega) following the manufacturer’s suggestion and 100 ng gDNA was used for qPCR. The cycle threshold (Ct) value was extrapolated to estimate the amount of human gDNA using a standard curve and then normalized to the total gDNA amount obtained from each organ/tissue. The male nuclear diploid genome spans for 6.27 gigabase pairs and weighs 6.41 pg (20). We calculated the number of cells by dividing the gDNA amount with 6.41 pg DNA/cell. Data from ten animals per group are presented as mean±SEM.
To assess the effect of 5-FC administration, MSCs/CD cells instead of naïve MSCs were injected to the right striatum as mentioned above. Then the animals were randomly assigned to two groups. 5-FC (Archimica) was orally given to one group at a dose of 1,000 mg/kg/day for a week. On day 0, 1, 3, 8, 14, and 28 after the transplantation, gDNA was isolated from the ipsilateral hemisphere from the animals with and without 5-FC and used for qPCR analysis. Data from 5∼7 animals per group are presented as mean±SEM.
Immunohistochemistry
The animals were deeply anesthetized with 2,2,2 tribromoethanol (200 mg/kg; i.p., Sigma-Aldrich), and then perfused transcardially with 10% neutral buffered formalin (BBC Biochemical). The brain was extracted, post-fixed in 10% neutral buffered formalin overnight at 4℃ and embedded in paraffin. The sections were prepared with 5 μm thickness. For immunostaining, antigens were unmasked by exposure to microwave radiation in 10 mM sodium citrate buffer (pH 6.0) and exposed to 0.3% H2O2 in distilled water for 30 minutes to block endogenous peroxidase activity. The sections were incubated with anti-human mitochondria antigen (hMT, mouse, 1:100; Millipore) and anti-Iba1 for microglia (rabbit, 1:3,000; WAKO) and then with biotinylated anti-mouse or -rabbit secondary antibodies (1:200; Vector Laboratories). After incubation with avidin-biotin complexes generated using a VECTA-STAIN ABC Kit (Vector Laboratories), immunoreactive proteins were visualized with 3,3’-diaminobenzidine (Sigma-Aldrich) as substrate to detect horse-radish-peroxidase activity. For fluorescence immunostaining, antigen retri-eval was performed by boiling with 0.05% citraconic anhydride in distilled water. The sections were incubated anti-CD antibody (CD, rabbit, 1:200; Young In frontier) and then with Alexa Fluor 488 or 568 conjugated secondary antibodies. Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining was performed using a In Situ Cell Death Detection Kit, TMR red (Cat. No 12156792910; Roche) following the manufacturer’s protocol. The sections were counter stained with Hoechst 33258, Pentahydrate (bis-Benzimide) (Cat. No H3569; InvitrogenTM) to show the nuclei. The sections were mou-nted and scanned at 20×resolution using a Scanscope CS digital slide scanner (Aperio Technologies) for bright images or Zeiss Axio Scan Z1 slide scanner (Carl Zeiss) for fluorescent images.
Statistical analysis
Statistical analyses were performed using SigmaPlot v14 software (Systat Software). Data were analyzed using the Student’s t-test or one-way ANOVA. Significant differences were further evaluated using Holm-Sidak method. p-value<0.05 was considered statistically significant. All data are expressed as the mean±SEM.
Go to :
Results
PK study of naïve MSCs in nude mice
We investigated PK aspect of human MSCs after local transplantation into a rodent model by qPCR analysis. To establish the standard curve for qPCR, reference samples (ranging 0.01∼100 ng) were prepared by serial dilution of human gDNA in mouse gDNA. The qPCR reaction was performed using human Alu-specific primers and then the standard curve was established using Ct values (Fig. 1A). The lower limit of quantification (LLOQ) was found 0.005 ng/ml in 3 sets of experiments with 5 replicates per assay. The slope and Y-intercept of the curve were −3.53±0.17 and 13.79±0.31, respectively. R2 value of 0.99 indicated the accuracy and precision of the assay were acceptable (Fig. 1B, 1C).
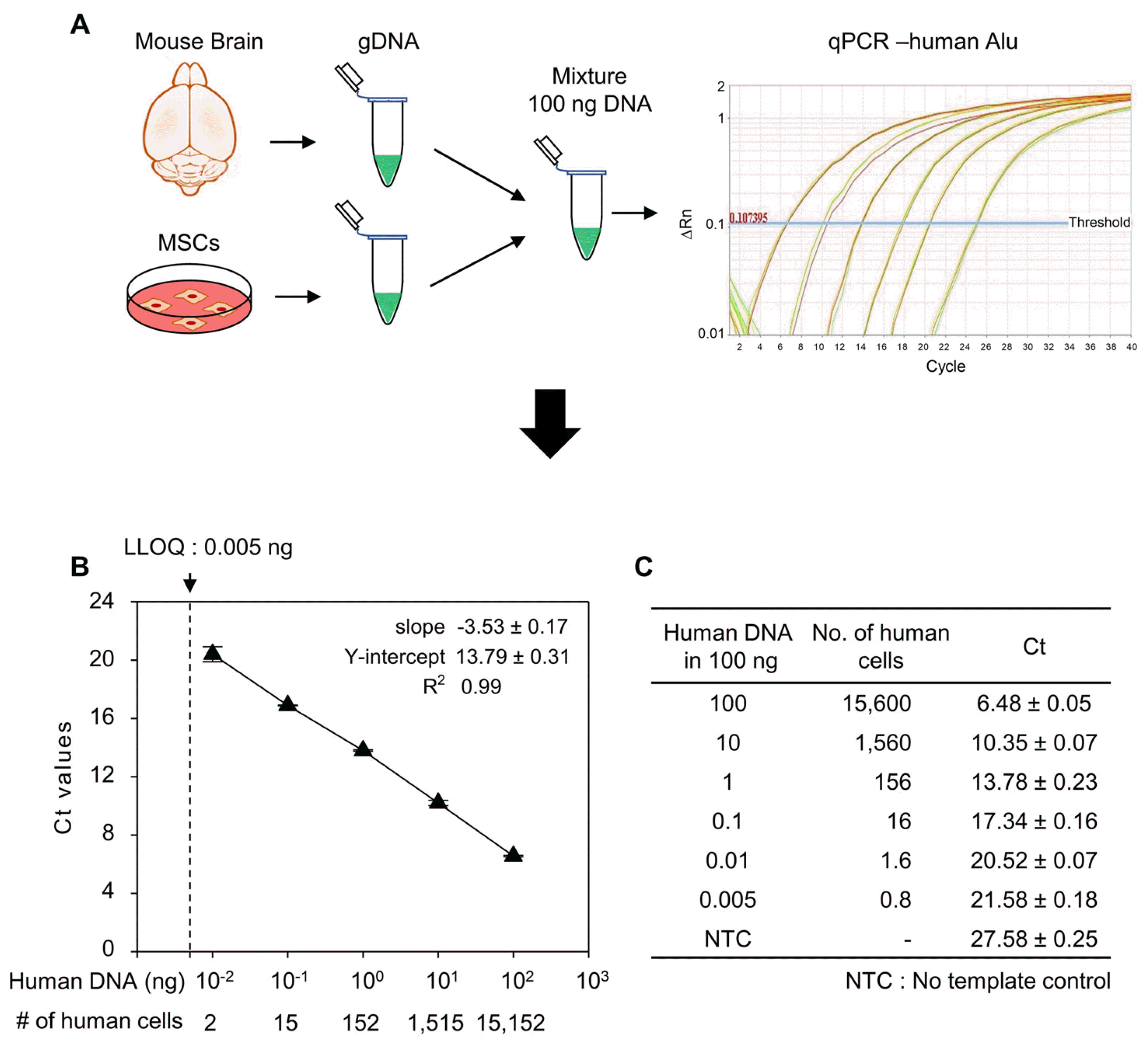 | Fig. 1Quantification of human cells by Alu J specific polymerase chain reaction (PCR) of genomic DNA (gDNA). (A) Schematic representation showing the method to prepare the human gene standard and human Alu J specific quantitative PCR (qPCR). gDNA from human mesenchymal stem cells (MSCs) were mixed with mouse brain gDNA to prepare standard DNA for qPCR. (B) Standard cu-rve generated from standard DNAs’ cycle threshold (Ct) values. (C) Con-centration of standard DNA used for Alu J qPCR and their respective Ct values obtained from qPCR. Data are mean±SD of three independent experiments. LLOQ: lower limit of quantification.
|
Next, we transplanted 3×105 MSCs in the brain of nude mice and extracted gDNA at different time points (0, 1, 3, 8, and 28-day) (Fig. 2A). The human Alu-specific signals were found in the ipsilateral hemisphere but not in the contralateral hemisphere. The signals declined substantially by 70% on day 3 and dropped below the LLOQ between day 8 and 28. The signals in the lung and liver were negligible at levels below LLOQ during the first week and were considered statistically insignificant (Fig. 2B). It is noteworthy that no signals were detected in the blood samples or any other organs (including spleen, lymph node, heart, kidney, pancreas, bone marrow, and gonads) throughout the entire period (data not shown). The results indicated that transplanted MSCs were confined to the injection site and did not migrate to the contralateral brain, lung, and liver. The data also suggest that disappearance of the xenografted human cells might be attributed to the residual immune response in the brain of nude mice.
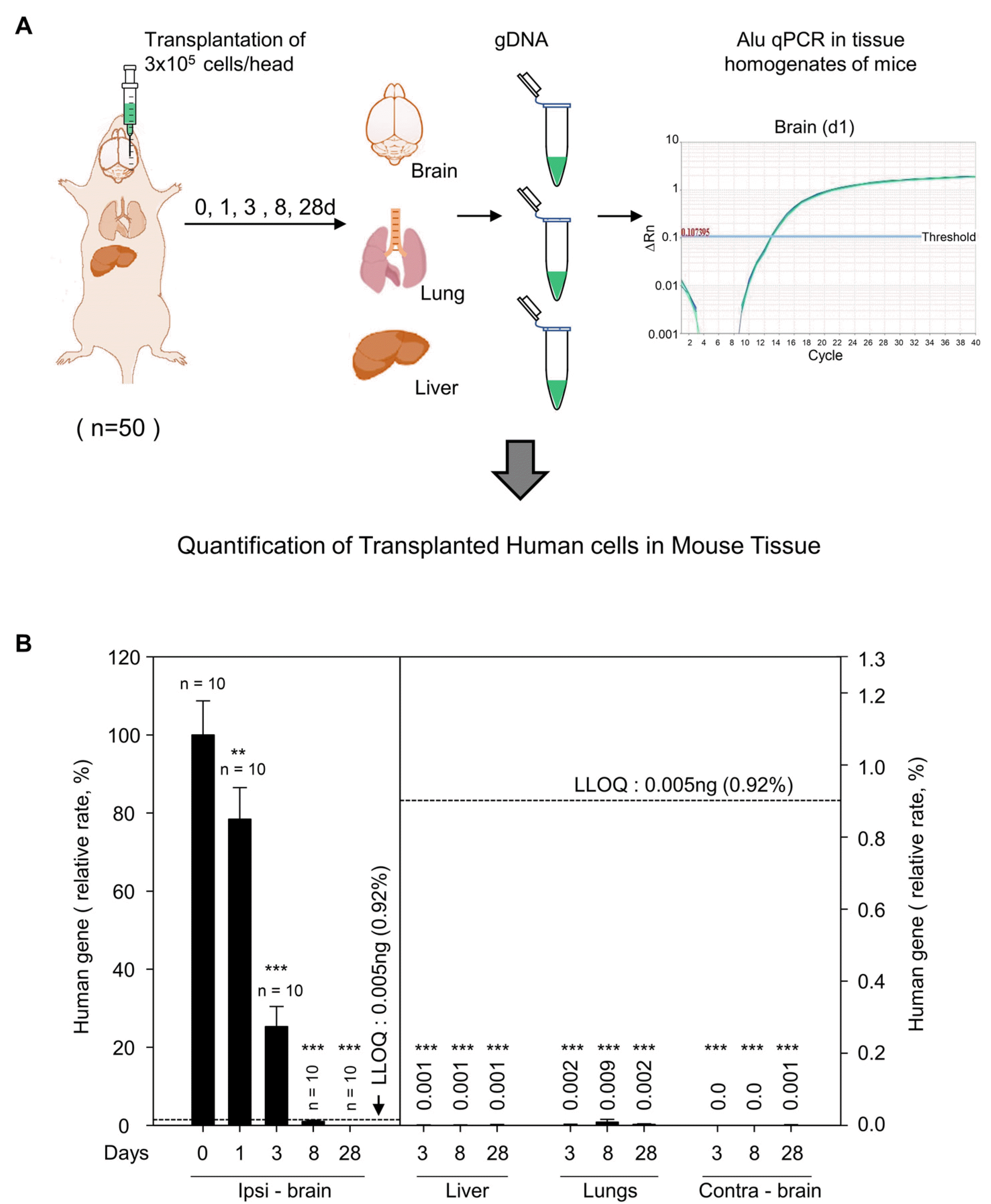 | Fig. 2Pharmacokinetics study of human mesenchymal stem cells (MSCs) transplanted into nude mice brain. (A) Schematic representation showing injection of MSCs in nude mouse striatum and genomic DNA (gDNA) preparation from brain, lungs, and liver on day 0, 1, 3, 8, and 28 after cell transplantation. (B) Quantification of human gene in ipsilateral brain (ipsi - brain), liver, lungs, and contralateral brain (contra - brain) from gDNA at indicated days by Alu J specific quantitative polymerase chain reaction (qPCR). Total number of animals per group is indicated just above bar graph in ipsi - brain. Low limit of quantification (LLOQ) is indicated by dotted lines. Values above bar graph in liver, lungs, contra - brain indicate relative rate of human gene in these organs compared to ipsi - brain at day 0. Data are mean± SEM of at least 10 animals per group. **p<0.01, ***p<0.001, compared to day 0, ipsi - brain group; one-way ANOVA test.
|
Effect of in vivo ablation of neighboring cells on transplanted MSCs/CD fate in the brain
In order to investigate the role of residual immune system in the xenograft model, we employed a chemical ablation method involving CD and 5-FC. We introduced a bacterial CD gene to MSCs to obtain MSCs/CD cells (Fig. 3A). MSCs/CD convert 5-FC to 5-FU that can interfere with DNA and RNA metabolism. Consequently, MSCs/CD undergo self-induced cell death while simultaneously inducing cell death to nearby bystander cells in the presence of 5-FC (Fig. 3A). The cellular properties of MSCs/CD cells were similar to those of naïve, unmodified MSCs. Both MSCs and MSCs/CD could undergo adipogenic, osteogenic and chondrogenic differentiation, as shown by Oil Red positive adipocytes, alizarin red-positive osteocytes, and Alcian blue-positive chondrocytes, respectively (Fig. 3B). FACS analysis indicated that surface antigens including CD29, CD90, CD105 (positive), CD34, CD45, and HLA-DR (negative) were similar in both cell types (Fig. 3C, 3D). These findings suggest that genetic modification with the bacterial CD gene do not alter the multilineage differentiation potential and surface antigenicity, which are the characteristics defined for MSCs by the International Society for Cellular Therapy (21).
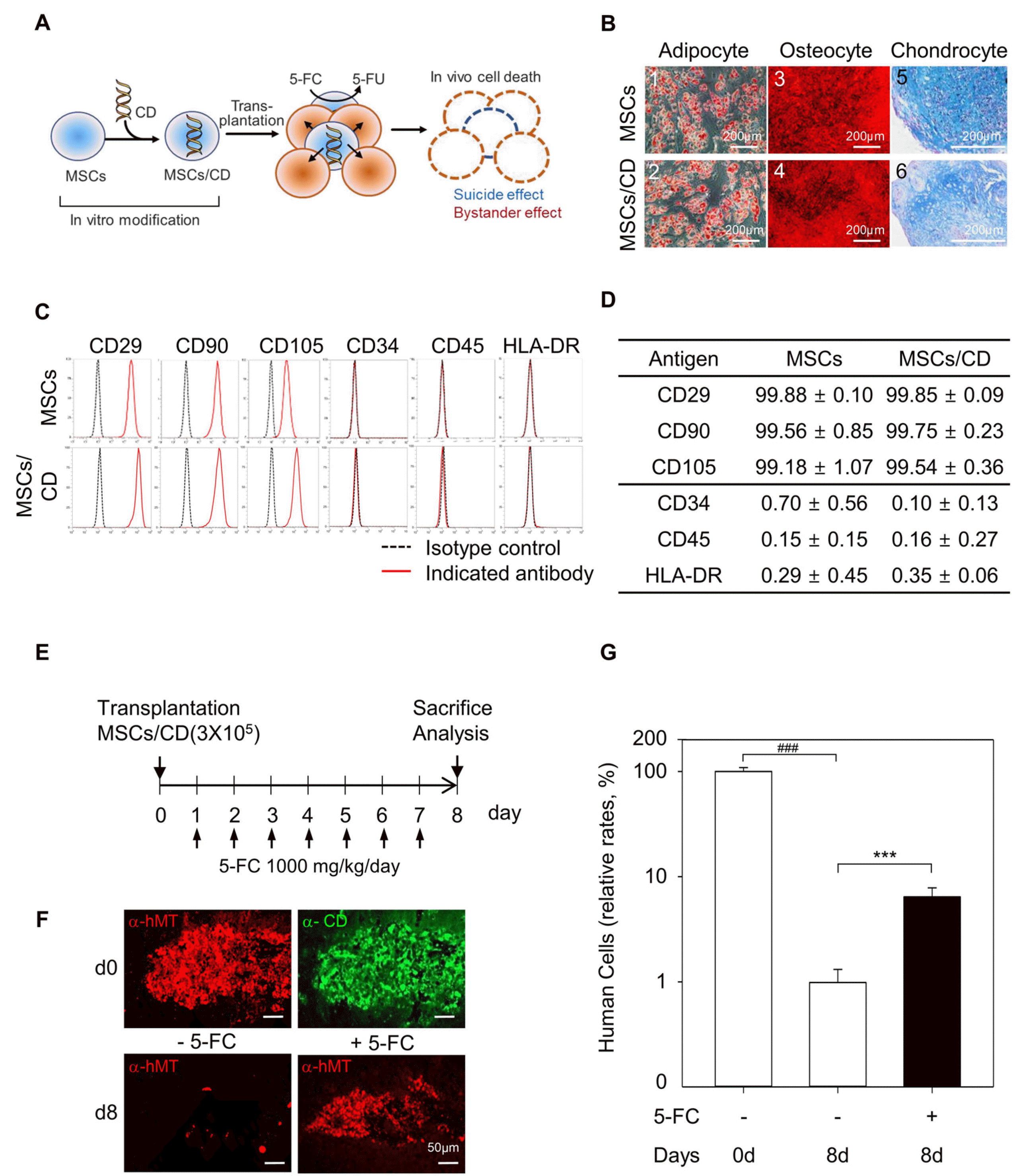 | Fig. 3Generation and characterization of mesenchymal stem cells expressing a cytosine deaminase (MSCs/CD), and effect of in vivo ablation of neighboring cells on the cell fate of transplanted MSCs/CD in the brain. (A) Schematic diagram of 5-fluorocytosine (5-FC) induced suicidal effect and bystander effect in MSCs/CD. (B) Mesodermal differentiation of MSCs and MScs/CD. Adipogenic differentiation showing Oil Red positive lipid droplets in bright field images of MSCs and MSCs/CD (B1, B2). Osteogenic differentiation showing alizarin red S stained precipitates in bright field images of MSCs and MSCs/CD (B3, B4). Chondrogenic differentiation of MSCs and MSCs/CD showing Alcian blue positive chondrocytes in bright field image (B5, B6). Scale bar=200 μm. (C) FACS analysis showing the surface antigen expression in MSCs and MSCs/CD. Isotype controls were used to determine the backgrounds. Black dotted peaks indicate the results obtained from cells stained with isotype control antibodies and red peaks indicate the results of cells stained with the indicated specific target antibodies. (D) Summary of FACS analysis showing the similar phenotype of MSCs and MSCs/CD: positive for CD29, CD90, and CD105 and negative for CD34, CD45, and HLA-DR. Data are mean±SEM from 3 independent experiments. (E) Experimental plan for 5-FC treatment to mice after MSCs/CD transplantation. (F) Representative immunofluorescence images show the detection of human mitochondrial-positive cells (α-hMT) with and without 5-FC administration. Scale bar=50 μm. (G) Comparison of relative rates of human cells detected at day 0 (d0) and day 8 (d8) with or without 5-FC administration. Data are mean±SEM of at least 10 animals per group. ###p<0.001, compared to d0 without 5-FC administered group; ***p<0.001, compared to d8 without 5-FC administered group; Student’s t-test.
|
After transplantation of 3×105 MSCs/CD in a similar manner, 5-FC was orally administered at a dose of 1,000 mg/kg/day for one week (Fig. 3E). The animals were sacrificed immediately after transplantation (day 0) and on day 8 for qPCR and immunohistochemical analyses. The animals that did not receive 5-FC administration were used as controls. Surprisingly, immunohistochemistry revealed that the presence of MSCs/CD was prolonged as seen by hMT- and CD-immunoreactivity at the injection site on day 8 (Fig. 3F). Consistently, the Alu signals robustly decreased on day 8 but persisted at a higher level with 5-FC administration than the control without 5-FC (Fig. 3G).
To further evaluate the effect of 5-FC, we transplanted MSCs/CD cells (3×105 cells) and randomly divided the animals into two groups on day 0. From the next day, 5-FC was orally given to one group at a dose of 1,000 mg/kg/day for one week and then the mice were sacrificed at various time points (day 0, 1, 3, 8, 14, and 28) (Fig. 4A). Genomic DNA was extracted from the ipsilateral hemisphere and subject to qPCR analysis. The remaining MSCs/CD cells were quantitated by qPCR in a similar manner shown in Fig. 1 (using the Ct values for human Alu-specific signal). The animals not receiving 5-FC were used as the controls. The number of MSCs/CD cells in both the control and 5-FC treated groups decreased significantly during the first week, but the rate of decline was slower with 5-FC treatment (Fig. 4B). Consistently, immunohistochemistry also revealed a prolonged presence of the hMT-immunoreactivity in the 5-FC treated group than in the control (Fig. 4C).
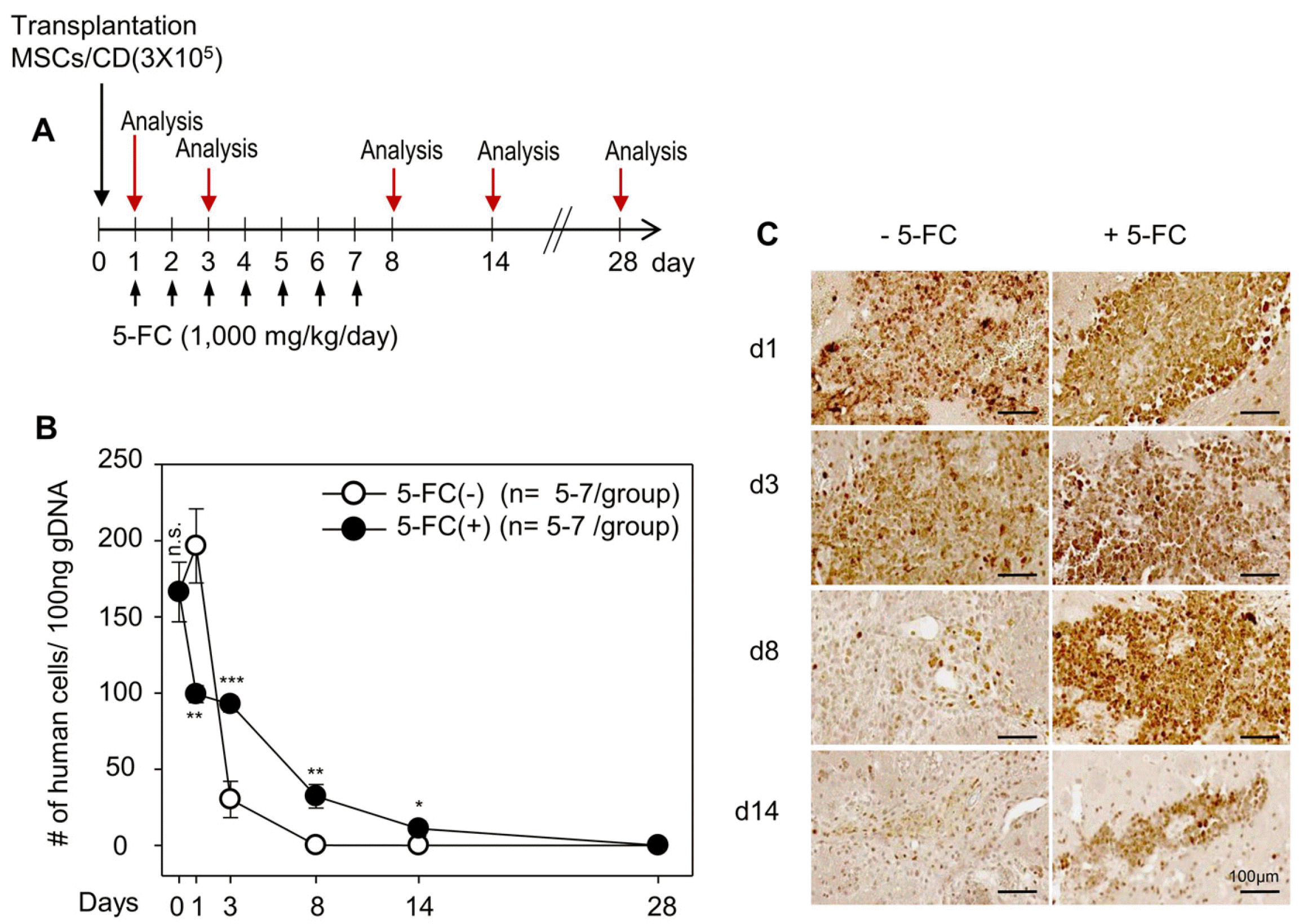 | Fig. 4
In vivo ablation of injected and neighboring cells with cytosine deaminase (CD) and 5-fluorocytosine (5-FC) prolongs the transplanted cells at the injection site. (A) Experimental plan for 5-FC administration (1,000 mg/kg/day) to nude mice brain after mesenchymal stem cells expressing a CD (MSCs/CD) (3×105 cells) transplantation. Animal were sacrifice and genomic DNA (gDNA) extracted from injected brain at day 0 (d0), d1, d3, d8, d14, and d28 after cell transplantation. The 5-FC non administered group was used as a control. (B) Data are mean±SEM of at least 5 animals per group. *p<0.05, **p<0.01, ***p<0.001, compared to data without 5-FC administered group; Student’s t-test. Comparison of human cells detected by quantitative polymerase chain reaction in the ipsilateral brain at d0, d1, d3, d8, d14, and d28 with or without 5-FC administration. (C) Representative immunohistochemical images with anti-human mitochondrial antigen (hMT) showing the human cells at the injection site of the animals. Scale bar=100 μm.
|
TUNEL staining revealed that the signal was evident from day 1 in the control group, suggesting that the transplanted cells underwent spontaneous cell death (Fig. 5A). The signal persisted until day 3 but disappeared by day 8. Interestingly, during day 3 to day 8, there was a significant increase in the Iba1-immunoreactivity, indicating robust activation of microglia (Fig. 5B). In contrast, in the 5-FC treated group, the TUNEL signal continued to intensify until day 8 (Fig. 5A), indicating MSCs/CD produce 5-FU in vivo. Simultaneously, 5-FU also induced cell death in resident microglia near the injection site through bystander activity, consequently suppressing Iba1-immu-noreactivity (Fig. 5B). Eventually the TUNEL-positive signals disappeared by d14 regardless of 5-FC treatment.
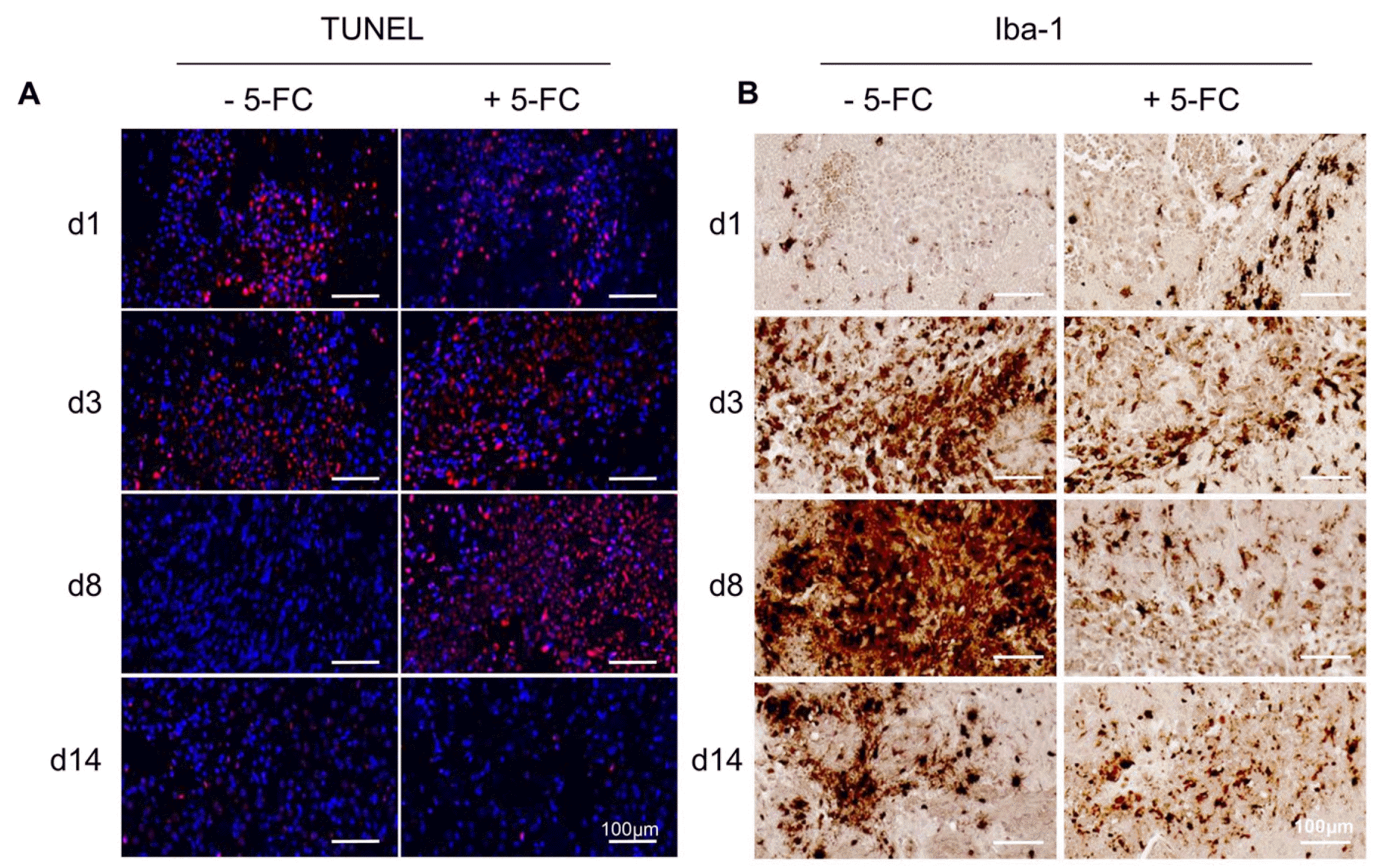 | Fig. 5
In vivo ablation of injected and neighboring cells with cytosine deaminase (CD) and 5-fluorocytosine (5-FC) differentially modulates immune response at the injection site. (A) Serially sectioned brains were stained for cell death at the injection site with Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL, red). The sections were counter-stained with Hoechst 33258 to show the nuclei (blue). The TUNEL-positive cells were detected at the injection site at day 1 (d1) in the control group, whereas they were dramatically increased at day 3∼8 (d3∼d8) in the 5-FC treated animal. (B) Serial sections were stained with an anti-Iba1 antibody to identify activated microglia at the injection site. Note dramatic increases of Iba-1 positive microglia only in the control group but not in the 5-FC treated group. Scale bar=100 μm. A minimum of two animals per group were utilized in our study. The representative one is shown.
|
Go to :
Discussion
Optimizing candidate selection for target therapeutic areas is an essential goal of preclinical PK and PD studies in drug discovery and development. While PD aspects of MSCs can depend on the inherent nature of therapeutic cells, their PK aspects is largely determined by the administration route. Unlike intravenous administration, the PK studies of MSCs after local transplantation is limited.
In this study, we investigated the biodistribution of xenografted human MSCs in a rodent model. To accurately quantify the remaining human MSCs in the animal, we employed three strategies. Firstly, we used immune compromised nude mice, which are commonly used for evaluating tumorigenicity due to their ability to facilitate efficient engraftment. Secondly, we targeted the brain as the transplantation site, taking advantage of its well-known immune privileged nature. Lastly, we implemented a qPCR method using human Alu-specific primers as a surrogate marker. This approach enabled us to precisely track and quantify the transplanted MSCs, providing an unbiased estimation of the remaining human cells within the mouse organ context.
The qPCR analysis revealed that the MSCs predominantly remained at the injection site within the ipsilateral hemisphere. However, their presence substantially declined over a week and eventually became undetectable after a month (Fig. 2B, 4C, without 5-FC). TUNEL staining revealed that these cells underwent cell death during day 1∼3 (Fig. 5A, without 5-FC) and then their presence became minimal at the injection site on day 8. Interestingly, this timeframe coincided with the peak activation of microglia, the residential immune cells in the brain (Fig. 5B, without 5-FC). The findings indicate that xenografted MSCs have a relatively short lifespan of less than a month due to the immune response elicited by the residual immune cells in the immune compromised animal. Further-more, the MSCs do not spread to other organs after local transplantation in the brain.
To investigate the role of the residual immune system, we employed a chemical ablation method using the CD enzyme capable of exerting both suicide and bystander effects (Fig. 3A). Both human-Alu specific signals and hMT-immunoreactivity persisted longer in the 5-FC treated group than in the control (Fig. 4B). This observation was reliable as two independent approaches consistently proved the presence of more hMSCs in the presence of 5-FC, although the TUNEL-positive signals were higher during day 3∼8 in the 5-FC treated group (Fig. 5A, compare without and with 5-FC). Interestingly, during this period, the activation of microglia remained minimal in the 5-FC group (Fig. 5B, with 5-FC). This finding suggests that microglia also undergo cell death via bystander functions of MSCs/CD, leading to delayed immune response to apoptotic MSCs/CD cells. This inverse correlation between the degree of remaining human cells and the activation of microglia suggests that the absence of microglia prolongs the presence of MSCs/CD in the 5-FC treated group. It should be noted that PK results were similar in animals with naïve MSCs and the animals with MSCs/CD cells without 5-FC in the control (compare Fig. 2B, 4B, without 5-FC), further validating that our approach of using qPCR analysis is reliable.
Nude mice have been extensively used in preclinical studies for evaluating the tumorigenic potential of stem cells and establishing xenograft tumor models for testing anticancer drugs. However, the findings of this study have limitations in terms of generalizing the biodistribution of MSCs in disease models. The biodistribution of MSC-based therapeutics in disease animal models may differ from what has been observed in normal animals, considering the high tropism of MSCs towards damaged tissues or cancer (22, 23).
Nevertheless, our study focusing on the PK of MSCs in the brain reveals potential benefits that can be translated to clinical applications. Recently, the CD gene has recently emerged as a promising therapeutic tool to treat the solid tumors in both preclinical and clinical studies (15, 24-26). However, the presence of suppressive immune cells, such as regulatory T cells, myeloid-derived suppressor cells, and tumor-associated macrophages, within the tumor microenvironment can hinder the effectiveness of anticancer therapies. Consequently, targeting these cells has become a major focus in cancer immunotherapy (27, 28). It would be interesting to investigate whether intra-tumoral transplantation of MSCs carrying the CD gene can modulate the suppressive immune cells in the vicinity, thereby enhancing the efficacy of anti-cancer treatments.
In conclusion, our study on the PK of MSCs in the brain provides promising results that have the potential to enhance therapeutic efficacy in clinical settings. Fur-thermore, our findings regarding immune modulation by MSCs/CD and 5-FC in the brains of nude mice offer valuable insights for optimizing therapeutic regimens involving the CD gene and 5-FC for solid tumors.
Go to :
 XML Download
XML Download