

 PDF
PDF Citation
Citation Print
Print



Introduction
The neural tube, an embryonic neural tissue, is a precursor of the central nervous system and is produced through a sophisticated developmental morphogenetic process called neurulation (1-3). Defects in the neurulation process often cause a developmental malformation called neural tube defects (NTDs), which is one of the most common birth defects, whose prevalence varies 0.5∼2 per 1,000 births depending on the country (4, 5). Although more than 400 genes are responsible for the failed neural tube closure process in mice (6), no conclusive evidence has determined the genetic factors responsible for human NTDs (7, 8). For genetic predisposition, women carrying affected fetuses have an empirical recurrence risk of 3% in subsequent pregnancies; however, this risk increases to approximately 10% after the birth of a second NTD embryo (5, 9). This emphasizes the importance of genetic factors; however, the influence of the environment on the development of NTD phenotypes cannot be ignored. Wide-ranging environmental exposures, such as air pollution, disease, nutrition, exposure to occupational chemicals and physical substances, and substance abuse, are the risk factors that increase the incidence of human NTDs (5, 10-12). Nevertheless, with some exceptions, majority of these chemicals failed to induce NTDs in animal models. Therefore, there is a great need for new experimental models for human NTDs to determine the susceptibility variables that define the gene–environmental interactions that contribute to the etiology of NTDs.
Neural organoids produced by human pluripotent stem cells (hPSCs) are promising models for NTDs in vitro. Recently, we have established human spinal cord organoids (hSCOs) that recapitulate neurulation-like morphogenesis, which can be utilized for the quantitative analysis of NTDs (13). This system is suitable for drug toxicological screening and includes two types of analysis: (1) bright field image-based observation for the progress of morphogenesis and (2) validation of final products based on neural tube structure. The morphological processes of hSCOs can be classified into three stages according to their morphological features: neural plate, neural folding, and neural tube. The initial screening of antiepileptic drugs that cause NTDs as side effects in hSCOs revealed that valproic acid (VPA) had the strongest association with the defects in tube morphogenesis in hSCOs. To enhance our understanding of the mode and molecular me-chanism of VPA-induced NTDs using an hSCO model, we determined the sensitive periods causing tube morphogenesis defects and examined transcriptome changes. Here, we proposed a molecular scenario wherein VPA treatment interfered with the polarized distribution of tight junctional complex proteins, thereby disrupting the apicobasal polarity of neuroepithelial (NE) cell sheets and eventually retarding neural tube folding morphogenesis.
Go to : 
Materials and Methods
Maintenance of human induced pluripotent stem cells
hiPSCs derived from epidermal fibroblasts were maintained on Matrigel (354277; Corning)-coated plates in mTeSR1 (Cat# 85850; STEMCELL Technologies). They were maintained in 5% CO2 at 37℃ with daily media changes and were passaged every 4∼7 days using ReLeSR (Cat# 05872; STEMCELL Technologies) into small clumps and replated onto precoated culture dishes. Experiments were performed on hiPSCs at less than 50 passages.
Generation of hSCOs
hSCOs were generated as the previously described protocol (13). Briefly, hiPSC clumps were plated on Matrigel-coated plates in mTeSR1. After 2 days, mTeSR1 was replaced with differentiation medium (DM) consisting of DMEM/F-12 (11320033; Life Technologies), 1% N2 (17502048; Life Technologies), 2% B27 (17504044; Life Technologies), 1% nonessential amino acids (11140050; Life Technologies), 1% penicillin/streptomycin (15140122; Life Technologies), and 0.1% β-mercaptoethanol (21985023; Life Technologies). SB431542 (10 μM, 1614; Tocris Bioscie-nce) and CHIR99021 (3 μM, SML1046; Sigma-Aldrich) were added to DM for 3 days. The differentiated colonies were dissociated with Accutase (07920; STEMCELL Tech-nologies) and seeded onto a 96-well low attachment plate (5,000 cells/well) in DM supplemented with basic fibroblast growth factor (bFGF) (20 ng/ml, 233-FB; R&D Systems). The hSCOs were fed daily for four days. On day 7, hSCOs were cultured in DM containing retinoic acid (0.1 μM) without bFGF for 8 days with daily medium change. To induce NTDs, hSCOs were cultured in DM containing VPA (1 mM, P4543; Sigma-Aldrich). Brightfield images were acquired using an EVOS microscope (Life Technologies) to quantify the morphogenetic processes.
Optical clearing and three-dimensional imaging
hSCOs were fixed with 4% paraformaldehyde (PFA; Biosesang) in PBS for 30 minutes, washed three times with PBST (0.1% Triton X-100 in PBS), and incubated with blocking solution (6% BSA, 0.2% Triton X-100, and 0.01% sodium azide in PBS) overnight. For three-dimensional (3D) whole-mount immunostaining, hSCOs were immersed in a primary antibody diluted in blocking solution for 48 hours. The antibodies used in this study were as follows: mouse anti-ZO1 (1:250, 33-9100; Invitrogen), rabbit anti-ZO2 (1:250, 71-1400; Invitrogen), and goat anti-SOX2 (1:250, sc-17320; Santa Cruz). The primary antibody was then washed with PBST three times for 10 minutes each. The samples were then incubated with the appropriate secondary antibody and Hoechst33342 diluted in the blocking solution for 48 hours. Subsequently, hSCOs were washed with PBST three times for 10 minutes each and mounted onto a cover glass (24×40 mm) with mounting solution (25% urea and 65% sucrose in H2O) for optical clearing. All steps were performed in a 0.2 ml micro-tube with gentle shaking at room temperature (RT). All images were acquired using a Leica TCS SP8 Confocal microscope.
Microarray
To analyze the changes in the transcriptome profile of hSCOs induced by VPA, samples were prepared using the following protocol: until day 11, the hSCOs were cultured in the same manner as the control group. On day 11, when the majority of hSCOs showed neural folding, the hSCOs-VPA group was cultured in DM containing 4 mM VPA. After 4 days, VPA-treated hSCOs were harvested, along with the control group. RNA purity and integrity were evaluated using an ND-1000 Spectrophotometer (NanoDrop) and Agilent 2100 Bioanalyzer (Agilent Technologies). The Affymetrix whole transcript (WT) expression array process was performed according to the manufacturer’s protocol (GeneChip WT PLUS Reagent Kit). cDNA was synthesized using the GeneChip WT Amplification Kit following the manufacturer’s instructions. The sense cDNA was then fragmented and biotin-labeled with terminal deoxynucleotidyl transferase using the GeneChip WT Terminal Labeling Kit. Approximately 5.5 μg of the labeled DNA target was hybridized to the Affymetrix GeneChip Human 2.0 ST Array at 45℃ for 16 hours. Hybridized arrays were washed and stained on a GeneChip Fluidics Station 450 and scanned on a GCS3000 Scanner (Affymetrix). Signal values were computed using Affymetrix GeneChip Com-mand Console software.
NTDs in vivo with VPA
Pregnant female (C57BL/6) mice were purchased from DAEHAN BIOLINK and VPA was purchased from Sigma-Aldrich (P4543). Pregnant female mice at 9 days post-coitus received freshly dissolved VPA (400 mg/kg) in saline three times (9 AM, 3 PM, and 9 PM) intraperitoneally (14). The control group received an equivalent volume of saline solution. Mice were sacrificed 12 hours after the last injection, and the dissected embryos were subjected to im-munohistochemistry.
Immunohistochemistry
Mouse embryonic tissue and organoid samples were fixed by immersion in 4% PFA overnight at 4℃ and washed several times in PBS. They were then incubated in 30% sucrose in PBS at 4℃ until completely submerged, embedded in Tissue-Tek “optimal cutting temperature” (O.C.T. Compound; Sakura-Finetek), frozen on dry ice, cryosectioned serially at 20 or 40 μm thickness and collected onto New Silane III coating slides (5118-20F; Muto Pure Chemicals Co. Ltd) (15). For immunostaining, samples were permeabilized with PBST (0.1% Triton X-100 in PBS) three times for 5 minutes each at RT, blocked with a solution (3% BSA and 0.2% Triton X-100 in PBS) for 30 minutes at RT, and then incubated with the respective primary antibody diluted in blocking solution overnight at 4℃. The antibodies used in this study were as follows rabbit anti-SOX2 (1:500, AB5603; Millipore), mouse anti-ZO1 (1:250, 33-9100), rabbit anti-ZO2 (1:250, 71-1400), mouse anti-occludin (1:200, 33-1500; Invitrogen), and rabbit anti-phospho-myosin Light Chain 2 (1:250, 3671s; Cell Signaling Technology). Samples were then washed with PBS three times for 5 minutes each at RT and then incubated with the respective secondary antibody and Hoechst33342 diluted in blocking solution for 30 minutes at RT. The secondary antibody was subsequently washed with PBST, and the samples were mounted on Crystal Mount (M02; Biomeda). Images were captured and processed using a Leica TCS SP8 Confocal microscope.
Statistical analysis
Statistical analysis were performed using unpaired Stu-dent’s t-test. All analysis were conducted using GraphPad Prism 9 software (GraphPad), and the results are presented as mean±SEM. Statistical significance was set at p<0.05.
Ethics approval and consent to participate
The human induced pluripotent stem cell (hiPSC) study was approved by the Institutional Review Board of Korea University (KUIRB-2020-0246-07). All animal maintenance and experimental procedures were approved by members of the Laboratory Animal Research Center at the Korea University College of Medicine (KOREA-2016-0194).
Go to :
Results
Modeling of VPA-induced NTDs with hSCOs
The duration of VPA treatment was divided into three stages depending on the status of morphological changes (stage I, before surface folding; stage II, near the onset of surface folding; stage III, near the termination of surface folding) if there are critical stages in the VPA-induced NTDs (Fig. 1A). Based on the bright-field images, alterations in the morphogenetic processes were verified (Fig. 1B), and quantified (Fig. 1C). Early treatment with VPA (stage I) appeared to cause a pause or delay in the progression of morphological processes, but washout of VPA after stage I resulted in delayed completion of folding mor-phogenesis. Treatment of VPA at stage II caused only marginal delay of the folding morphogenesis, and treatment at stage III did not significantly alter the folding morpho-genesis. This suggests that the VPA reversibly delays but not permanently disrupts the folding morphogenesis.
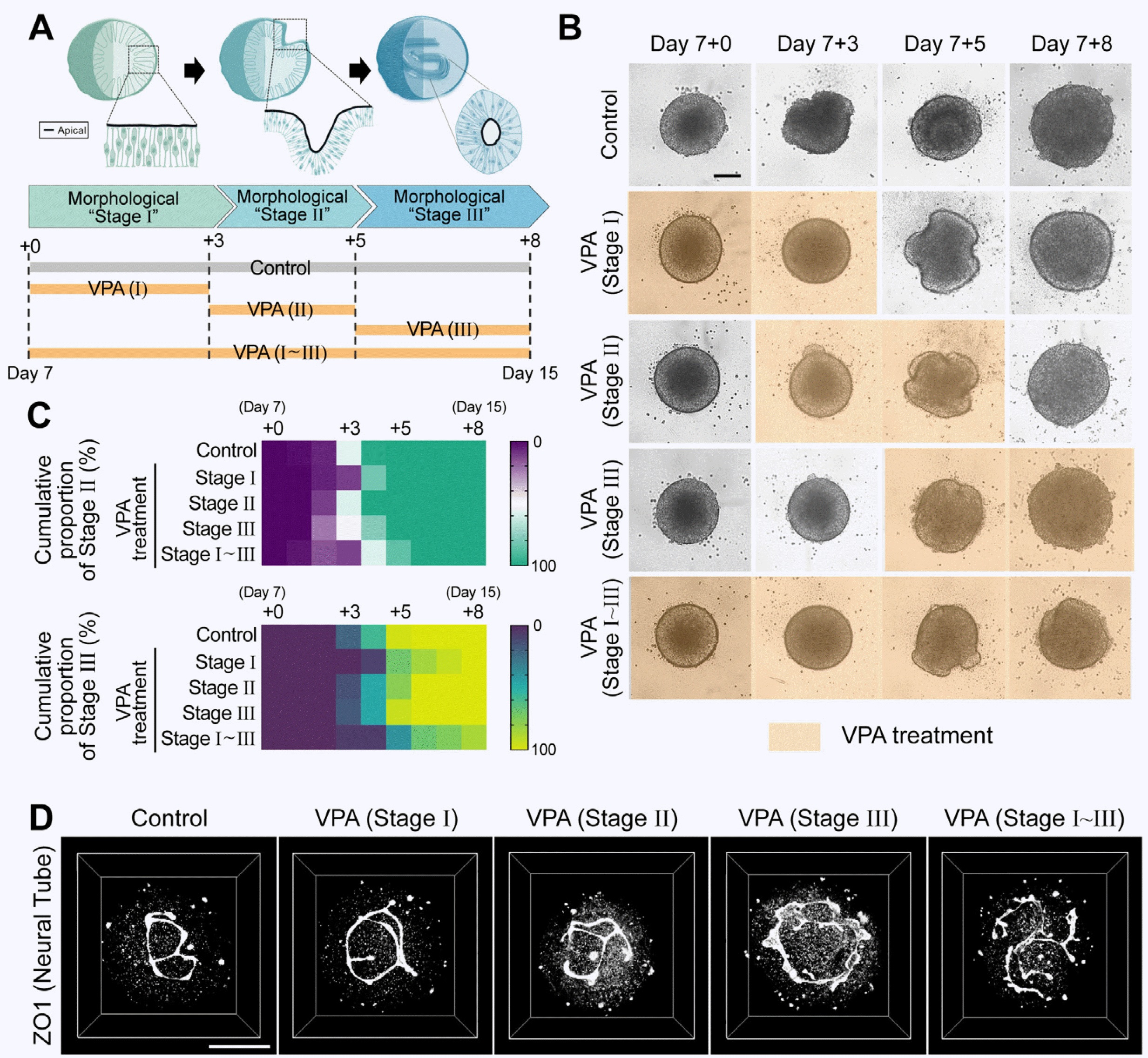 | Fig. 1Modeling of valproic acid (VPA)–induced neural tube defects with human spinal cord organoids (hSCOs). (A) Experimental model to study the effect of duration of VPA treatment on morphogenesis. (B) Re-presentative time-lapse images of neural tube morphogenesis in VPA-treated groups compared with the control. Each image was acquired using an automated imaging system. Scale bar= 200 μm. (C) Quantification of morphogenesis with different durations of VPA treatment. The color of each box indicates the cumulative proportion of neural folding (upper) or neural tube (bottom). Stage of hSCOs at the indicated culture time. (D) Three-dimensional neural tube morphology with different VPA treatment durations on day 15. The structure was visualized using an apical marker and ZO1 staining. Scale bar=100 μm.
|
Previously, we observed that VPA also induces opening or incomplete closure of the neural tube structure, even after the termination of gross folding morphogenesis. There-fore, high-resolution 3D imaging of apical ZO1 was performed to examine the detailed structures of the interna-lized neural tube in hSCOs on day 15 (Fig. 1D). The tube structures in the VPA treatment groups at stage I or II appeared to be virtually normal and exhibited closed neural tube structures. In contrast, the VPA treatment group at stage III exhibited an open neural tube structure, similar to the VPA stage I∼III group (Fig. 1B, 1C). In summary, VPA exposure at an early stage caused reversible and delayed folding morphogenesis, while the late stage (stage III) appeared to be sensitive to VPA on internalized tube closure morphogenesis.
Upregulation of cell–cell junction-related genes in VPA-treated hSCOs
To explore the molecular changes induced by VPA, we performed a microarray experiment and compared the gene expression profiles of hSCOs with or without VPA treatment. A scatter plot demonstrated that in VPA-treated samples, 432 genes were upregulated and 279 genes were downregulated significantly with a fold change≥2 on the log2 scale compared with the control (Fig. 2A). To further explore the biological functions of the upregulated differentially expressed genes (DEGs), we performed gene ontology (GO) enrichment analysis. Interestingly, the upregulated DEGs were involved in GO terms associated with cell junction organization, including “bicellular tight junction assembly”, “apical junction assembly”, “and cell–cell junction assembly” (Fig. 2B). Further, we identified networks of interacting proteins associated with the upregulated DEGs using the GeneMANIA database (Fig. 2C) (16). Interaction network analysis indicated that cell–cell adhesion related genes, particularly tight junctions, were primarily involved in one interaction network and formed a compact cluster (Fig. 2C).
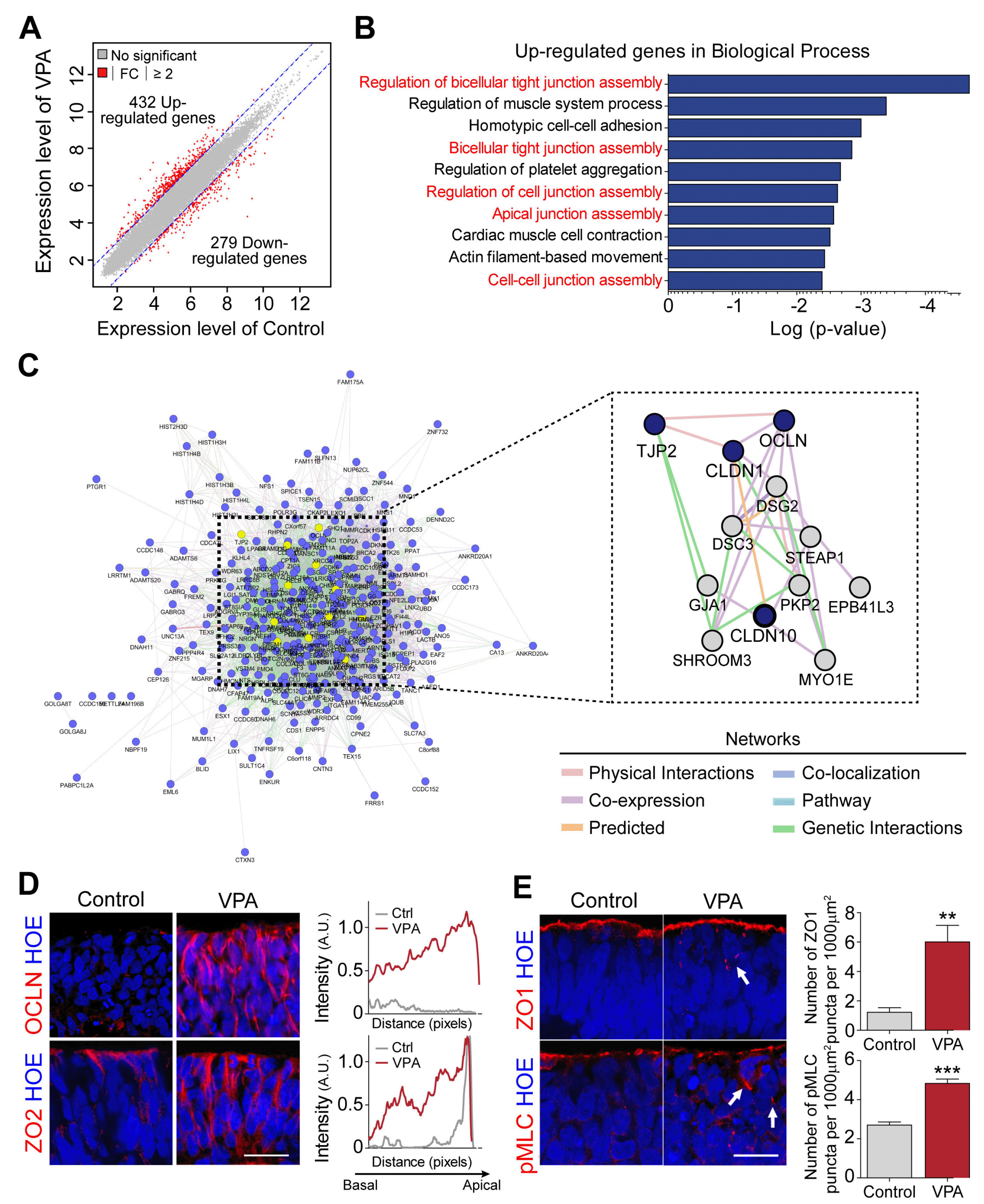 | Fig. 2Transcriptome analysis in human spinal cord organoids (hSCOs) exposed to valproic acid (VPA). (A) Scatter plots of transcriptome levels in hSCOs treated with VPA compared with the controls. Red circles denote the genes whose expression levels were significantly altered (≥2-fold) in VPA-treated vs control groups. (B) Biological processes associated with significantly upregulated genes. (C) Network plot of the upregulated genes enriched in VPA-treated hSCOs. Interactive network is generated using GeneMANIA plugin in Cytoscape. (D) Immunohistochemical evaluation of the upregulation of tight junction proteins, Occludin (OCLN), and ZO2. Average intensity of tight junction proteins was measured along the apico-basal axis. Scale bar=20 μm. (E) Perturbation of polarized localization of ZO1 and pMLC proteins in the neuroepithelial cells of VPA-treated hSCOs. White arrows indicate the ectopic expression of proteins in the VPA-treated group. Bar graphs show the number of ectopic ZO1 and pMLC punctas in neural-plate layers. Unpaired Student’s t-test was used for comparing two groups (**p<0.01 and ***p<0.01). Scale bar=20 μm. FC: fold change, Ctrl: control.
|
To verify the significance of the above bioinformatic analysis, we examined the distribution of two tight junction-related proteins, Occludin and ZO2, whose expression was identified as a VPA-induced gene group (Fig. 2D). These two proteins were substantially upregulated in VPA-treated neural tubes in hSCOs. More importantly, these proteins were spread across all NE cells, while they were more constricted to the apicolateral domains of the NE cells in the control. Furthermore, markers for apicobasal polarity, pMLC, and ZO1 were often misplaced in the non-apicobasal domains, suggesting that apicobasal polarization of VPA-treated NE cells was disrupted (Fig. 2E).
Effect of tight junction proteins expression and distribution in VPA-induced NTDs
Next, we aimed to correlate the disruption of tight junctions with tube defects in various VPA-treated groups. Interestingly, VPA, at any single stage, was sufficient to enhance ZO2 misplacement (Fig. 3A, 3B). Considering that folding morphogenesis is a reversible process, the removal of VPA could resume morphogenesis, suggesting that ZO2 induction was not tightly linked to folding morphogenesis. On the other hand, significant misplacement of ZO1 was seen in the VPA (stage I∼III)-treated group alone (Fig. 3C, 3D). Therefore, tight junction defects appeared to have a greater role in inducing, whereas the role of individual proteins such as ZO2 misplacement was not essential.
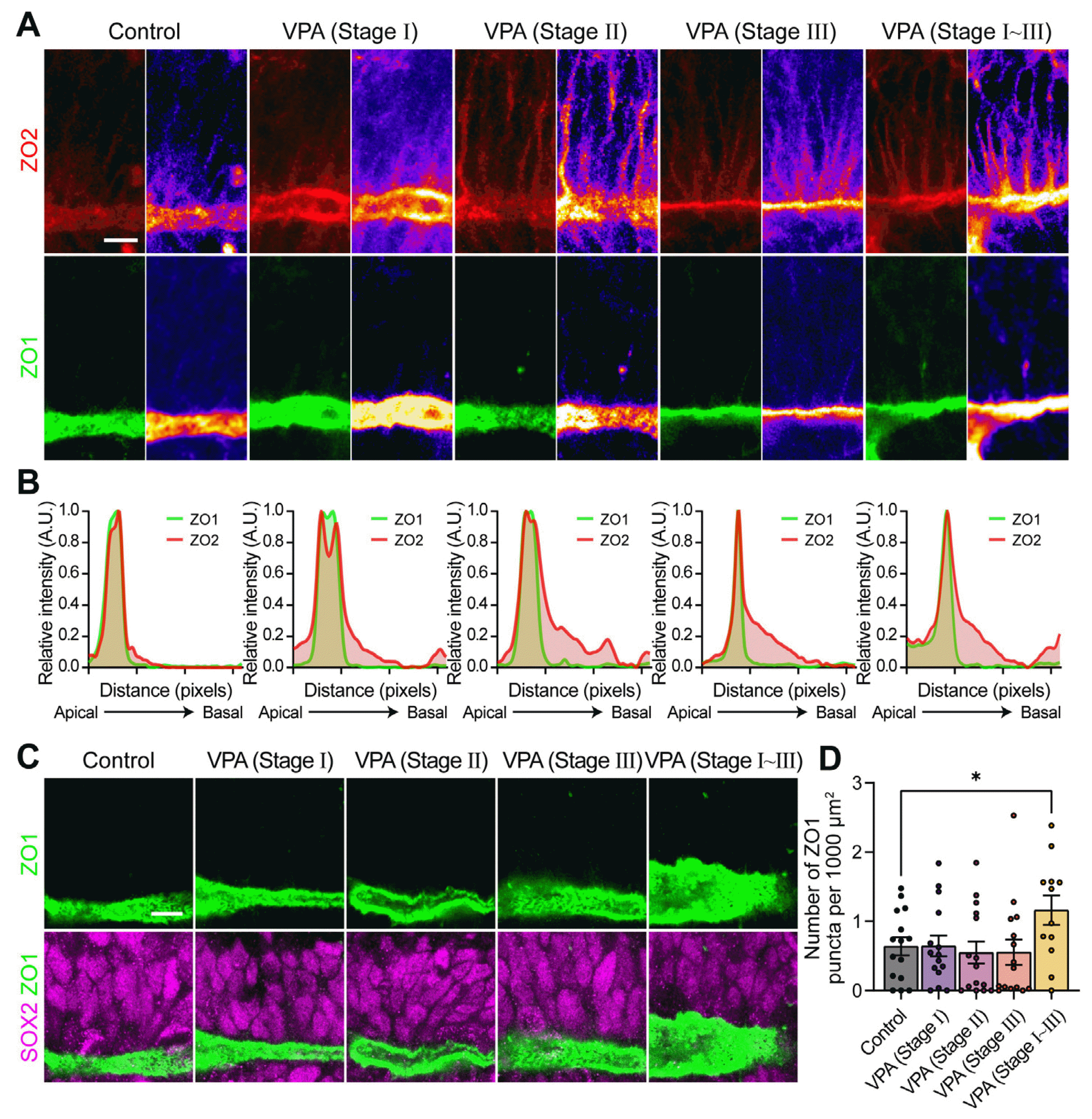 | Fig. 3Effect of tight junction proteins expression/distribution in valproic acid (VPA)-treated human spinal cord or-ganoid. (A) Immunohistochemistry of upregulation of tight junction proteins, ZO2 (red) and ZO1 (green) caused by VPA treatments for different durations. Scale bar=10 μm. (B) Line plots denoting the intensity of ZO1 and ZO2 staining along the closed/opened neural tube. (C) Pertur-bation of polarized localization of ZO1 proteins in neuroepithelium. The neuroepithelial cells were identified as SOX2 (magenta) staining and cellular arrangement surrounding apical marker, ZO1 (green). Scale bar=10 μm. (D) Quantification of the number of ectopic ZO1 punctas within neuroepithelial cell layers. Bar graphs represent mean±SEM, with all the points plotted. Unpaired Student’s t-test was used for comparing two groups (*p<0.05).
|
Effects of VPA treatment in vivo using mouse model
Finally, we investigated whether VPA affected neural tube formation in a mouse model with tight junction defects. Previously, it was reported that VPA treatments to the mouse embryos at E9 can cause the NTD-like malformations (17, 18). The embryos treated with VPA displayed closed neural tubes consisting of Sox2-labeled neural progenitors, but their apicobasal polarity was disrupted. Specifically, the F-actin signal, which is normally concentrated at the apical side, expanded towards the lateral/basal sides, and punctas of pMLC were observed on the non-apical side (Fig. 4A). Interestingly, these embryos also showed abnormal distribution of Zo1 and increased expression of Zo2, similar to what is observed in the hSCOs model. However, the expression of occludin was not significantly altered, suggesting that the regulation of these genes in response to VPA may depend on the species (Fig. 4B). In addition, misplacement of tight junction markers was evident in VPA-treated embryos, suggesting that the disruption of tight junctions is potentially the main phenomenon that is tightly linked to the development of NTDs.
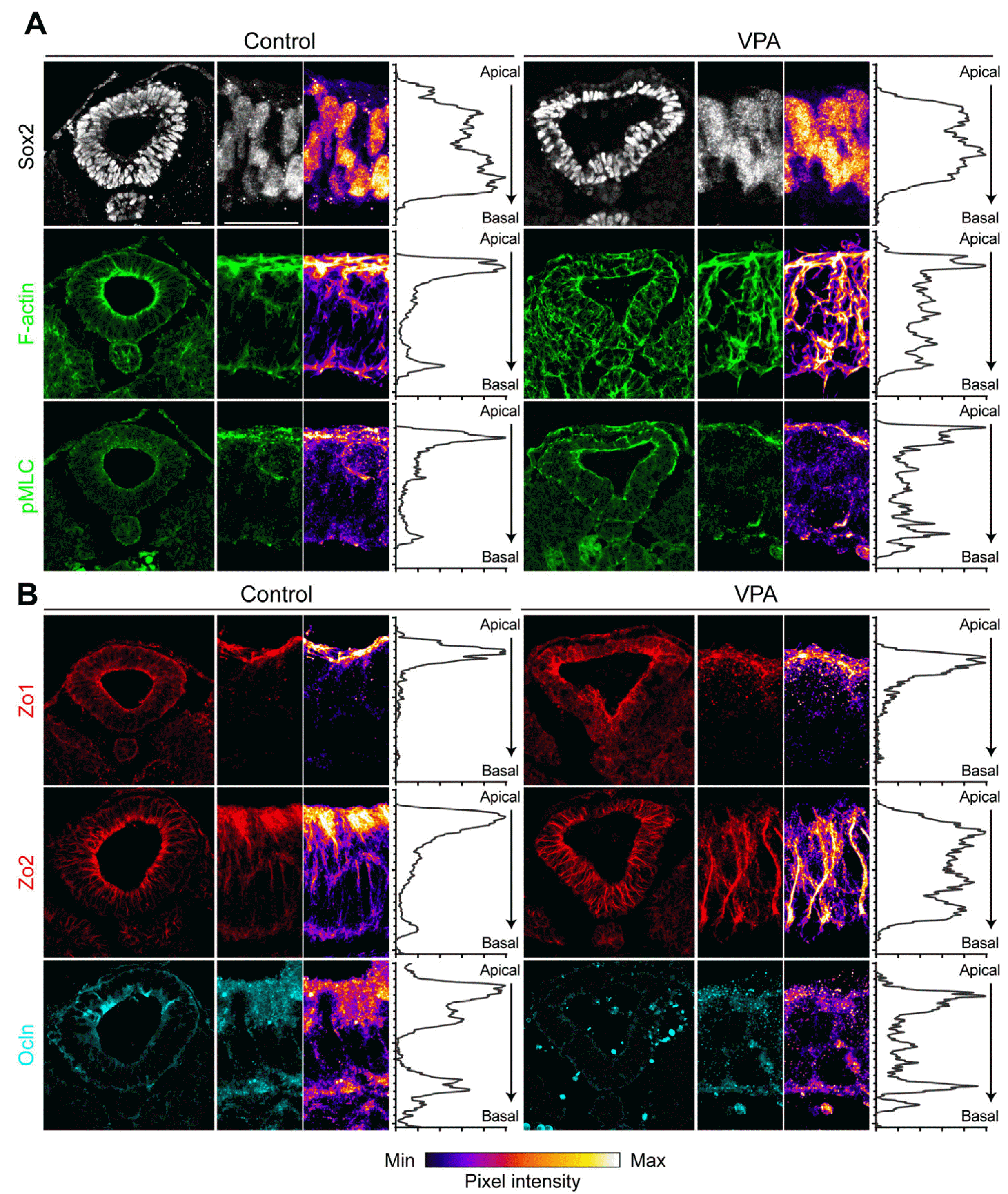 | Fig. 4Effect of valproic acid (VPA) on neural tube in the mouse em-bryo. Immunohistochemistry of VPA-induced neural tube defect mouse models. Various markers were used to observe changes in the expre-ssion/distribution of actin filaments (A, F-actin and pMLC) and tight junction proteins (B, Zo1, Zo2, and Ocln). Dissolved VPA was administrated three times to pregnant female mice at the time of primary neuru-lation. Scale bars=20 μm. Min: minimum, Max: maximum.
|
Go to :
Discussion
The 3D hSCO model system was used in this study to recapitulate neural tube morphogenesis, allow for quantitative analysis of the procedure, and show morphological defects in response to VPA exposure. Based on these characteristics, we investigated the etiology of VPA-induced NTD. Exposure to VPA alters the cell–cell junctional organization, particularly the aberrant expression of tight junction proteins. These features were observed in both the human organoid and mouse embryo models.
These results highlight the advantages of utilizing organoids as a model system to understand the underlying mechanisms of human diseases. Compared to the in vivo model, the organoid model has the advantage of being more accessible and allowing for a more creative experimental design, while avoiding ethical issues. It is challenging to identify which morphogenic phase might be affected by risk factors because neural tube morphogenesis involves a continuous process in vivo. From this perspective, the hSCO model appears to provide an appropriate experimental model for investigating these and the related issues. We subdivided and examined the morphogenetic stage-dependent deficiencies induced by VPA exposure in organoids using a previously established quantitative analysis system. Early stage VPA exposure caused delays or interruptions in morphogenic procedures, but recovery after VPA removal was reversible. Late-stage VPA exposure results in more vulnerable responses than those at earlier stages. Interestingly, our discovery that VPA causes disruption in the later stages of neural tube closure is consistent with the previous data reported using mice models (18). According to Hughes et al. (18), VPA exposure disrupts spinal neural tube closure via a biomecha-nical effect, particularly in the posterior neuropore in the caudal part (“Closure 5”). However, we also observed difference in the alteration of the expression pattern of the tight junction protein occludin between hSCOs and mice in response to VPA treatments (Fig. 4). Additionally, there are significant differences in the genetic risk factors associated with NTDs between humans and mice, providing further evidence of interspecies differences in the processes of neural tube closure (6, 8). These interspecies differences in the developmental process underscore the importance of using in vitro human experimental models.
With the advancement of stem cell/organoid technology, various in vitro models have been developed to recapitulate human neural tubes (19-22). In adherent culture systems, micropatterning of hPSCs has been primarily used to provide geometric constraints and trigger morphogenesis (23-26). Furthermore, 3D neural rosettes generated by single lumen formation in the extracellular matrix have also been utilized for neural tube modeling, although it does not reflect comprehensive folding morphogenesis (27-29). Since VPA is one of the most well-known risk factors, several studies have attempted to induce NTDs by VPA exposure in in vitro as well as in vivo models. Although the results for VPA exposure in each model varied, VPA acted strongly on majority in vitro models and exhibited distinct defect phenotypes, such as inverse bending (24), inhibition of NE cell proliferation (25), and impediment of lumen formation (27).
There are several hypotheses on how VPA causes NTDs, including alterations in folate metabolism, increase in reactive oxygen species, and epigenetic dysregulation (30-34). For instance, the effect of VPA as a folic acid antagonist is primarily supported by a comparative analysis between the effects of methotrexate and VPA (35-37). This is also supported by the observation that folic acid rescued VPA-induced defects in zebrafish embryos as well as in in vitro neural tube models (27, 38). However, the precise molecular mechanisms underlying this phenomenon remain unclear. In this regard, our transcriptome studies suggest that tight junction defects may be closely associated with VPA-induced NTDs. The concept of changes in tight junctions caused by VPA exposure can be extended to a broader concept, such as disruption of apical protein complexes. This hypothesis is supported by previous reports of impaired cytoskeletal function and apical polarization following VPA treatment (18, 27). This is because tight junctional proteins are located on the apical domain of epithelial cells and interact closely with cytoskeletal elements, including actin filaments and micro-tubules. Consistent with this, Zhang et al. (27) proposed that VPA impedes lumen morphogenesis by altering cytoskeletal function and cell polarization. Furthermore, folate deficiency-induced NTDs are associated with the dysregulation of cytoskeletal components (39, 40). Collectively, these results suggest that VPA-induced NTDs are associated with apical protein assembly, particularly in tight junctions.
Go to :
 XML Download
XML Download