

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Methylation is an important molecular regulatory mechanism that can influence gene expression in addition to genetic, transcriptomic, and proteomic alterations.12 Methylation analysis involves determining whether specific regions of DNA sequences are methylated, such that methyl groups (CH3) are attached to them.1 Brain tumors are divided into various types and subtypes, each of which may have distinct methylation patterns.345 Analyzing these methylation patterns can help identify the characteristics of the tumor and assist in classification and prognostic predictions.
DNA methylation serves as a consistent biomarker and indicates the specific tumor cell type from which it originates. Large-scale examination of DNA methylation revealed unique patterns of each tumor.6 By employing machine learning techniques to analyze DNA methylation, it became possible to classify brain tumors that pose diagnostic challenges due to their unusual histopathology or diverse molecular genetic profiles.7 Additionally, these techniques have the potential to unveil previously unrecognized subtypes of brain tumors. Methylation profiles have been shown to redefine previously existing brain tumor type and subtype more clearly,6 for example, astroblastoma, MN1-altered.8 The discovery of new types and subtypes of brain tumors occurred through the characterization of an unmatched group of tumors, using a methylation classifier.39 These newly identified tumor types and subtypes have been incorporated into the 5th edition of the World Health Organization (WHO) classification of central nervous system (CNS) tumors. Examples of these newly included tumors are polymorphous low-grade neuroepithelial tumor of the young (PLNTY),10 high-grade astrocytoma with piloid features (HGAP),11 diffuse pediatric-type high-grade glioma, isocitrate dehydrogenase (IDH)-wildtype and H3-wildtype,12 infant-type hemispheric glioma,13 diffuse glioneuronal tumor with oligodendroglioma-like features and nuclear clusters (DGONC),14 and CNS neuroblastoma with forkhead box R2 (FOXR2)-activated,15 etc.
DNA methylation analysis has shown promising early results in the clinical application to CNS tumors and sarcomas.616 As a result, further advancements have been made in utilizing this approach in various other areas, including oral carcinoma, bladder cancer, breast cancer, ovarian cancer, and several other types of cancers.171819202122 Currently, the methods for detecting and categorizing cancers based on the methylation profile of cell-free DNA obtained from blood samples or cerebrospinal fluid are still in the early stages of development.23242526 The increasing advancements and broadening scope of DNA methylation research have enabled the identification of shared DNA methylation profilings among the same types of cancer. As a result, DNA methylation profiling has become an important auxiliary component of cancer diagnostic criteria and is now a important research conducted at prominent medical institutions like the German Cancer Research Center (Deutsches Krebsforschungszentrum, DKFZ)6 or National Cancer Institute (NCI) of USA.27
The Methylation Classifier developed by the Department of Neuropathology at DKFZ is a technology designed for molecular classification of brain tumors (https://www.molecularneuropathology.org/mnp). This classifier focuses on analyzing the methylation status of genes to accurately identify the type and classification of brain tumors.6 The DKFZ Methylation Classifier is a powerful tool to enhance patient care by precise pathological diagnosis and enabling personalized therapeutic approaches. It is continually refined and updated (now v12.8) as new insights and ensuring its accuracy and relevance in the field of neuropathology. However, the reference data (the methylation profiles of each brain tumor) is not fully accessible to researchers or external hospitals, which poses a significant obstacle to the utilization of methylation classifiers.
WHAT IS THE METHYLOME?
The methylome refers to the overall pattern of DNA methylation within an organism’s genome.28 DNA methylation is a chemical modification that involves the addition of a methyl group to the DNA molecule, typically occurring at cytosine residues within a CpG dinucleotide context, where a cytosine is followed by a guanine.
DNA methylation is an essential epigenetic modification that plays a crucial role in regulating gene expression and maintaining genome stability.1 It is also involved in various biological processes, including embryonic development, genomic imprinting, X-chromosome inactivation, and suppression of transposable elements.29
The methylome can vary between different cell types and developmental stages, as well as in response to environmental factors and disease states.30 Aberrant DNA methylation patterns have been associated with various diseases, including cancer,31 neurological disorders,32 and autoimmune disorders,33 etc.
The implementation of methylome analysis in classification of brain tumors
DNA methylation patterns can help identify different type and subtypes of brain tumors.6 For example, gliomas, which are the most common type of brain tumors, can be classified into different subtypes based on their DNA methylation profiles. These subtypes have been found to have different clinical features, treatment responses, and prognoses.34
These markers can be used to develop diagnostic and prognostic tests that detect the preexisting and novel brain tumors and differentiate them from other subtype of brain tumors.15 Aberrant DNA methylation can lead to the silencing of tumor suppressor genes or the activation of oncogenes, contributing to tumor development and progression. By understanding the methylome landscape of brain tumors, researchers can identify specific genes or pathways that are dysregulated and may serve as targets for therapeutic interventions.3536 Monitoring changes in the methylome during treatment can be used to assess treatment effectiveness and detect the emergence of treatment-resistant clones.3738
Typing and subtyping of brain tumors using methylation profile
Integrating methylome data with other molecular and clinical information can improve the accuracy of brain tumor classification and facilitate personalized treatment approaches.
In the realm of diagnostics, it is important to recognize that DNA methylation analysis does not provide a definitive solution for all diagnostic challenges. Its interpretation should be carried out within the context of the particular case at hand. Supplementary information, such as histopathology, immunohistochemistry, clinical data, and genomic analysis, continues to hold significant value in achieving an integrated diagnostic approach. It is crucial to understand that DNA methylation analysis is not intended to supplant the process of histologic interpretation. Nevertheless, the application of methylation profiling enables enhanced diagnostic precision.
Followings are several instances of molecular subtyping of brain tumors using methylation profile analysis.
Medulloblastoma
Despite various suggestions for additional subtypes of medulloblastoma by different research groups,3940 the 5th edition of the WHO classification of CNS tumors has officially recognized and accepted four major genetically defined subtypes of medulloblastoma,4 which are WNT-activated, Sonic hedgehog (SHH)-activated, groups 3 and 4 (Table 1).41
Table 1
A list of tumors for which methylation profiling proves useful in molecular subclassifications
WNT-activated subtype is characterized by the activation of the WNT signaling pathway, which plays a crucial role in normal brain development. WNT-activated medulloblastomas typically have a good prognosis and are associated with older age at diagnosis and accounting for approximately 10% of cases.42 Molecular alterations of WNT subtype usually have mutation of CTNNB1, DDX3X, SMARCA4 and TP53, and monosomy 6.43
SHH-activated subtype is driven by the activation of the SHH signaling pathway. SHH-activated medulloblastomas are the most common subtype in infants and young children and accounting for approximately 30% of cases. This subtype usually has mutations in the PTCH1, SMO, SUFU and TP53, amplification of GLI2 & MYCN, 3q gain, and 9q, 10q, and 17p loss.43 They have variable outcomes depending on specific genetic alterations and other factors. This group is further subdivided based on various characteristics including mutations in the TP53 gene. TP53-mutant has a worse prognosis compared to TP53-wildtype.4244
Group 3 subtype is characterized by aggressive tumor behavior and poor prognosis and accounting for approximately 20% of cases. Mutational profile of the group 3 usually includes GLI1, GFL1B activation, MYC and OTX-2 amplification, SMARCA4 mutation, i17q, gain of 1q, 7, and 18, and loss of 10q, 11, 16, and 17p.4145
Group 4 subtype is the most common molecular subtype of medulloblastoma, accounting for approximately 40% of cases. Group 4 can harbor KDM6A mutation, SNCAIP duplication, CDK6 amplification, and MYCN amplification, i17q, 7q gain, and 18q gain, 8p loss, 11p loss, and X loss. It is generally associated with an intermediate prognosis.3943
The molecular subtyping of medulloblastoma presents several challenges due to the complexity and epigenetic changes, such as super-enhancer hijacking without genomic changes.41 Molecular subtyping can be possible with integrating data analysis from multiple sources, such as transcriptome-based gene expression,46 genetic mutations,4041 and DNA methylation.44 Integrating and harmonizing these different types of data can be complex, and specialized tools and algorithms are required for accurate and reliable classification. Among them, subtyping by methylation is the most valuable if we classify medulloblastoma into four subtypes. The t-SNE plots of our hospital cases of medulloblastoma subtypes using methylation profile produced by illumina EPIC850K methylation microarray data are shown in Fig. 1.
Fig. 1
t-SNE clustering was applied to the CNS tumor methylation data obtained from the SNUH, along with reference methylation data from the German Cancer Research Center (DKFZ). The reference data used in this study was publicly available and obtained from the GSE90496 dataset. (A) Figure illustrates the t-SNE visualization of the methylation classes of various SNUH tumors, represented by different colors, in comparison to the DKFZ reference methylation classes of brain tumors, also represented by different colors. (B) Figure depicts the visualization of the SNUH data represented by the brown color, overlaid on the reference data from DKFZ, which are represented by the gray color. (C-E) Figures exhibit the t-SNE visualization of the methylation classes of specific tumor types, namely EPN, MB, and AT/RT, respectively.
t-SNE = t-distributed stochastic neighbor embedding, CNS = central nervous system, SNUH = Seoul National University Hospital, DKFZ = Deutsches Krebsforschungszentrum, MB = medulloblastoma, AT/RT = atypical teratoid/rhabdoid tumor, EPN = ependymoma, PFA/PFB = posterior fossa group A/B, SPNE = spinal ependymoma, MPE = myxopapillary ependymoma, TYR = tyrosinase-activated, MYC = MYC-activated, WNT = WNT-activated, SHH = Sonic hedgehog-activated, G3 = group 3, G4 = group 4, SHH INF = infantile sonic hedgehog-activated medulloblastoma.

Despite these challenges, ongoing research efforts are focused on addressing these difficulties and improving the accuracy and clinical utility of molecular subtyping in medulloblastoma.47
Ependymoma
Ependymoma is a type of brain tumor that arises from ependymal cells lining the ventricles of the brain and the central canal of the spinal cord. Ependymoma subclassified into supratentorial ependymoma (ST-EPN), posterior fossa ependymoma (PF-EPN), and spinal ependymoma (SP-EPN) by tumor location, molecular genetic changes and DNA methylation profile.48
ST-EPN primarily occurs in the cerebral hemispheres of the brain. It is further divided into two molecular groups, ST-EPN-ZFTA fusion-positive and ST-EPN-YAP1-positive: ST-EPN-ZFTA is characterized by a genetic alteration involving the ZFTA gene, most commonly a ZFTA::RELA fusion, but rarely MAML2 or YAP1 as the partner genes.49 It is associated with younger age at diagnosis, worse prognosis, and increased resistance to standard therapies. ST-EPN-YAP1 is characterized by genetic alterations involving the YAP1 gene. The most common partner gene is MAMLD1.48 It generally has a better prognosis compared to ST-EPN-ZFTA.
PF-EPN occurs in the posterior fossa region of the brain, which includes the cerebellum. It is further classified into two molecular subgroups, PF group A (PFA) and PF group B (PFB).45 PFA-EPN show no genetic abnormalities but have epigenomic changes including EZHIP overexpression and H3K27me3 loss.4550 It is associated with younger age at diagnosis, highly proliferative histopathology and poor prognosis. PFB-EPN is characterized by low-grade histopathology and generally has a better prognosis compared to PFA-EPN, but no significant genomic change.51
SP-EPN occurs in the spinal cord and both low-grade and high-grade SP-EPN are associated with neurofibromatosis type 2 or sporadic NF2 mutation or deletion.4952 However, ZFTA::YAP1 fusion EPN has been reported in the spinal cord, which are associated with high-grade molecular features and aggressive biological behavior.53
MYCN-activated spinal ependymoma is characterized by the activation or amplification of the MYCN gene.54 This subtype is rare, often present as the multiple tumors involving the multiple levels of the spinal cord and shows aggressive behavior.
These molecular subtypes of ependymoma provide valuable insights into the biology of the tumor, prognosis prediction, and potential treatment strategies, however, for molecular subtyping, we requires specialized techniques and equipment for genetic analysis, such as fluorescence in situ hybridization (FISH), reverse transcription-polymerase chain reaction (RT-PCR), or next-generation sequencing (NGS).55 These techniques may not be readily available in all clinical or research settings, limiting the widespread adoption and application of molecular subtyping.
Harmonizing classification systems and establishing consensus guidelines can help minimize these discrepancies. Methylation-based classification can overcome these complex problems, if reference data of the methylation profile of each subtype of ependymoma are available.
Atypical teratoid/rhabdoid tumor (AT/RT)
AT/RT is a rare and aggressive form of brain tumor that primarily affects infants and young children. Notably biallelic mutation of SMARCB1 or rarely SMARCA4, or partial or whole loss of chromosome 22.56 This gene is responsible for regulating the activity of other genes involved in cell growth and division. In AT/RT, the loss of SMARCB1 leads to uncontrolled cell growth and the development of tumors.
However, these tumors demonstrate an extremely simple cancer genome with a lack of additional mutation. Further analyzing tumor cells’ genetic and molecular features classifies AT/RT at the molecular level, enhancing disease comprehension and identifying treatment targets.57 A 2020 international consensus was documented for molecular classification of AT/RT, which permitted AT/RT-TYR-activated, ATRT-SHH-activated, and AT/RT-MYC-amplified, based on the nomenclature proposed by Ho et al.56 and Johann et al.58 Compound genomic and transcriptome study can subclassify, but methylation profile is the most convincing tool to subclassify AT/RT. Tyrosinase, melanoma-oncogene (microphthalmia-associated transcription factor, MITF), bone morphogenetic protein (BMP) pathway (e.g., BMP4) and developmentally related transcription factors such as orthodenticle homeobox 2 (OTX2) are highly overexpressed in AT/RT-TYR subtype. Whole or partial loss of one copy of chromosome 22 is accompanied by inactivating mutation in SMARCB1 on the other allele. Clinically, AT/RT-TYR patients represent the youngest with median age of 12 months (range, 0–108 months).
Most AT/RT-SHH cases show compound heterozygous point mutations of SMARCB1, while SMARCB1 homo- or heterozygous deletions are less common.56 The patients of this subgroup a more intermediate age group (median age, 20 months; range, 0–96 months), with patients on average younger than AT/RT-MYC patients and older than AT/RT-TYR patients. The supratentorial localization is more common than infratentorial. AT/RT-SHH subgroup splits up in 2 subtypes associated with either a mainly supratentorial location (AT/RT-SHH-1) or a mainly infratentorial (AT/RT-SHH-2) location by further DNA methylation analysis.56 Notch and SHH pathways genes are overexpressed in this subtype.
The AT/RT-MYC was named based on MYC oncogene overexpression. Some of HOXC cluster genes are overexpressed driven by H3K27-acetylation. A homozygous, broad loss of SMARCB1, covering several hundred kilobases, leads to SMARCB1 inactivation, but point mutations of this gene are rare in this subtype. Median age of the patients of this subtype is significantly higher than the other 2 subtypes (median, 27 months; range, 0–190.9 months). A majority of AT/RT-MYC arise supratentorial (50%) but all spinal and most adult AT/RTs including sellar AT/RT were MYC subtype in one cohort.5659
Additionally, ongoing research in this field aims to identify novel molecular targets that can be exploited for the development of more effective therapies for AT/RT. We can achieve this complex subtyping using methylation profiles.
THE USAGE OF A METHYLATION CLASSIFIER IN BRAIN TUMORS OF “NOT OTHERWISE SPECIFIED (NOS)” OR “NOT ELSEWHERE CLASSIFIED (NEC)” TYPE OR BRAIN TUMOR WITH DIAGNOSTIC DILEMMA
NOS or NEC brain tumors are often challenging to classify using traditional histopathological methods alone, as they may display atypical or ambiguous histopathological or molecular genetic features.4 A methylation classifier can provide valuable information about the molecular characteristics of these tumors, allowing for more accurate classification and potentially guiding treatment decisions.3 By comparing the methylation profile of a NOS or NEC tumor to a reference database of known tumor type and subtypes, the methylation classifier can help assign the tumor to a specific molecular subtype or provide insight into its closest molecular match. This can aid in determining the tumor's origin, behavior, and potential treatment options.
In gliomas, methylation profiling has enabled the identification of specific types, such as IDH-mutant gliomas (astrocytoma and oligodendroglioma) or glioblastoma, IDH-wildtype with specific molecular features; receptor tyrosine kinase 1 (RTK1), RTK2, and mesenchymal, primitive neuronal component subtype (https://www.molecularneuropathology.org/mnp/classifiers/14). Recently, novel Oligodendroglioma, IDH-mutant subtype is documented by DKFZ MC version 12.8. In cases where diagnosis is challenging based solely on morphological features, tumors like high-grade astrocytoma with piloid characteristics,6061 pediatric-type diffuse high-grade glioma without IDH and H3 mutations,1262 DGONC,1463 myxoid glioneuronal tumor, spinal ependymoma with MYCN amplification,5464 and CNS neuroblastoma with FOXR2-activated1565 can receive diagnostic assistance from a methylation classifier. In Table 1, we have compiled a list of brain tumors that can benefit from the use of a methylation classifier for diagnostic purposes. Additionally, Table 2 provides a compilation of brain tumors where the methylation classifier can aid in molecular subtyping.
Table 2
A list of tumors that methylation profiling is helpful for molecular subclassifications
DETECTION OF COPY NUMBER ABERRATIONS AND MGMT METHYLATION STATUS BY METHYLATION CLASSIFIER
“Copy number aberration” may increase (copy number gain) or decrease (copy number loss) the amount of DNA in a particular region of the genome. This can result in disease, including many forms of cancer. For example, glioblastoma, IDH-wildtype represent EGFR amplification, PTEN deletion, or chromosome 7 gain/10 loss.4 Oligodendroglioma has chromosome 1p- and 19q-codeletion. Those are molecular genetic signatures of glioblastoma and oligodendrogliomas.
“MGMTp methylation status” refers to the epigenetic alteration of the MGMT gene, which is a DNA-repair gene. Methylation is a process where a methyl group is added to DNA, which can change the activity of a DNA segment without changing its sequence. When the MGMT gene is methylated, it cannot properly repair damaged DNA.
The methylation classifier can elucidate the CNV and MGMT promoter methylation status. Fig. 2 demonstrates an example of copy number aberration, while Fig. 3 illustrates the methylation status generated by the methylation classifier. Both figures display four subtypes of medulloblastomas from Seoul National University Hospital.
Fig. 2
Examples of copy number aberration plots of four subtype of medulloblastoma generated from methylation 850K Epic microarray data. The first two plots are derived from the WNT-subtype, the next two plots are from SHH-activated group, followed by two plots from group 3, and the last two plots represent group 4 medulloblastoma.
SNUH = Seoul National University Hospital, SHH = Sonic hedgehog.
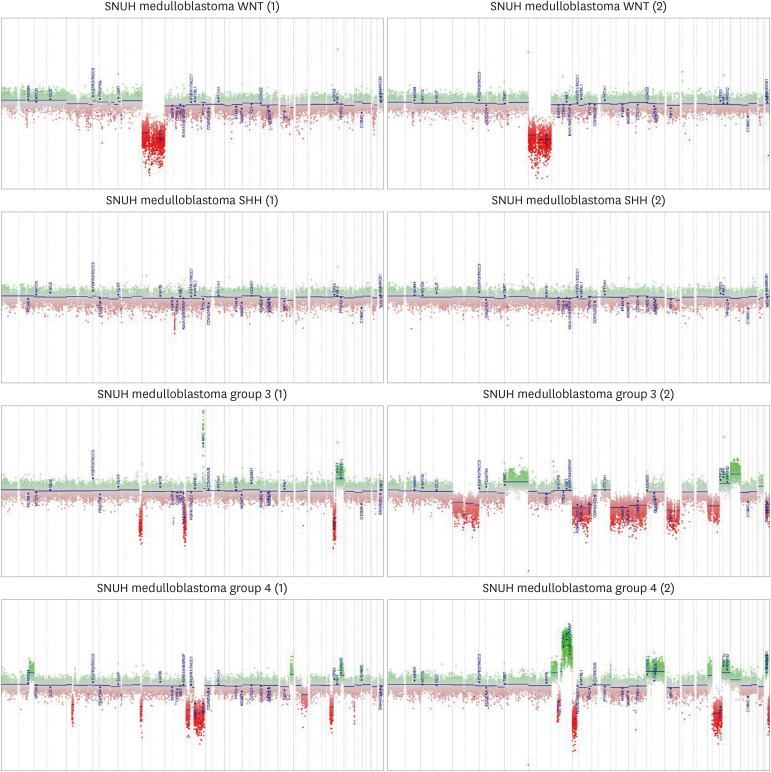
Fig. 3
Examples of MGMT promoter (MGMTp) methylated status predicted by DKFZ classifier with Illumina Epic850K methylation microarray data. The cutoff of methylated and unmethylated status is the red line (score = 0.3582). The results of MGMTp status, determined through methylation-specific polymerase chain reaction, align perfectly with the findings mentioned above. (A, B) glioblastomas, (C, D) oligodendrogliomas.
DKFZ = Deutsches Krebsforschungszentrum, SNUH = Seoul National University Hospital, IDH = isocitrate dehydrogenase.

ALGORITHM TO CLASSIFY BRAIN TUMORS BY METHYLATION PROFILE
In the realm of methylome analysis for brain tumor classification, several algorithms have been developed and employed. These algorithms typically involve a series of computational steps to extract meaningful features from DNA methylation data and classify brain tumors based on these features.
The process begins with the preprocessing of raw DNA methylation data obtained from tumor samples. This initial step includes quality control measures to ensure the reliability of the data, removing technical biases and filtering procedures to exclude any unreliable data points.
Once the data is preprocessed, the next step is feature selection or extraction. This involves identifying relevant features from the methylome data that are informative for tumor classification. Various techniques can be applied for this purpose, including differential methylation analysis to identify differentially methylated regions between tumor samples and control samples.22 Dimensionality reduction methods such as principal component analysis or feature selection algorithms like Least Absolute Shrinkage and Selection Operator identify the most informative features associated with tumor classification.318
The subsequent step is the construction of a classification model using machine learning or statistical algorithms, including support vector machines, random forests, logistic regression, or neural networks. The choice of algorithm depends on the nature of the dataset and the specific goals of the analysis. The model is trained using a training set consisting of labeled samples with known tumor types. Then the classifier has to be evaluated using appropriate evaluation metrics such as accuracy, sensitivity, specificity, or the area under the receiver operating characteristic curve.6
Finally, integrating the methylome data with other omics data, such as gene expression or genomic alterations, can further enhance the understanding of brain tumor biology.
THE LIMITATION OF BRAIN TUMOR CLASSIFICATION BY METHYLATION STUDY
The limitations of brain tumor classification through methylation studies are summarized in Table 3. The major obstacle lies in the fact that methylation classifiers are predominantly research-based, making them unavailable for patient charge. Thus, medical insurance does not provide coverage for methylation studies. Furthermore, in Asian countries where equipment and reagents need to be imported entirely, the costs incurred are more than three times higher than Western countries.
Table 3
The limitations of methylation-based classification in brain tumors

Moreover, the availability of comprehensive and diverse datasets poses a challenge. Methylation studies rely heavily on robust and extensive datasets encompassing a broad range of tumor types, subtypes, and clinical parameters. However, the acquisition of such datasets, with adequate sample sizes and comprehensive clinical annotations, can be inherently challenging. The scarcity of high-quality datasets may limit the generalizability and reproducibility of methylation-based classification models, hindering their translation into clinical practice. Particularly, in the case of recently recognized novel subtypes, these data are not publicly available in open databases, and inquiring authors of original papers or the DKFZ for such novel data yields scarce instances of provision.
Foremost the primary limitations lies in the potential for complexity of the epigenetic landscape. Methylation patterns, while insightful in discerning molecular subtypes, may not capture the full spectrum of heterogeneity present within brain tumors.66 This arises due to the existence of various epigenetic mechanisms beyond DNA methylation, such as histone modifications, chromatin remodeling, and non-coding RNA regulation, which collectively contribute to the intricate gene expression patterns observed in tumor cells.67 Consequently, the sole reliance on methylation patterns may overlook pertinent information essential to an overall classification scheme.
Another notable limitation pertains to the dynamic nature of the epigenome. Epigenetic modifications can exhibit temporal and spatial heterogeneity, influenced by various factors, including tumor microenvironment, therapy-induced changes, and clonal evolution.6668 Consequently, a single methylation snapshot may not fully capture the dynamic and evolving nature of brain tumors over time, potentially limiting the accuracy and reliability of classification efforts based solely on methylation profiles.
Inherent technical limitations associated with methylation profiling techniques must be acknowledged. These techniques, such as microarray-based or next-generation sequencing approaches, often exhibit inherent biases and limitations, including batch effects, variability in probe coverage, and sensitivity to DNA quality and quantity.69 Such technical limitations can introduce potential confounders and may impact the accuracy and robustness of methylation-based classification models.
CONCLUSION
In conclusion, while methylation studies have proven to be fairly valuable tools for brain tumor classification to guide treatment adjustments and help optimize therapeutic strategies, it is essential to acknowledge and address the limitations inherent to this approach. Recognizing the complex nature of the epigenome, the dynamic changes over time, the availability of comprehensive reference datasets, and the technical limitations of methylation profiling are crucial steps towards refining and augmenting the accuracy and clinical utility of methylation-based brain tumor classification.
 XML Download
XML Download