

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Various histological neoplasms originate in the pineal region. Pineal parenchymal tumors (PPT) are a rare group of tumors originating from pineal parenchymal cells. They account for less than 0.5% of central nervous system neoplasms and 20%–30% of tumors in the pineal region [12]. PPT are classified into three types: pineocytoma, pineal parenchymal tumor of intermediate differentiation, and pineoblastoma. Pineocytoma (PC) is a low-grade tumor that exhibits benign cytologic features [3]. Pineoblastoma (PB) is a malignant embryonal tumor that is prevalent in children and young adolescents [4]. Pineal parenchymal tumor of intermediate differentiation (PPTID) was first defined by Schild et al. [5] and classified by the World Health Organization (WHO) in 2000 [6]. It is characterized by intermediate morphological features and clinical behavior between PC and PB [7]. PC and PPTID are more common in adults than in children.
PPT can present with various symptoms associated with increased intracranial pressure. Common symptoms and signs are caused by hydrocephalus, brainstem compression, and cerebellar dysfunction [28]. The diagnosis and treatment of PPT pose challenges because of their location within the brain and proximity to deep cerebral veins, which can make surgical removal difficult [9]. The difficulty in choosing optimal treatment and predicting prognosis is due to the lack of clear-cut histological criteria and rarity of these tumors. We investigated the clinical data from patients of all ages with PPTs from a single institution to identify differences in clinical features, biological behavior, therapeutic strategies, and prognosis based on the histopathological grade. This study aimed to provide a comprehensive perspective on the spectrum of these rare tumors.
MATERIALS AND METHODS
Patient selection and clinical data
This study was approved by the Institutional Review Board (IRB No. 2102-122-1198) of Seoul National University Hospital (SNUH). The requirement for informed consent was waived due to the retrospective nature of the study. We reviewed medical records of 48 patients diagnosed with PPT at SNUH between 1990 and 2020. We excluded 3 patients who underwent initial surgery for diagnosis in other hospitals. The 2021 WHO classification shows five types of PPT [10]. We excluded papillary tumors of the pineal region (PTPR) and desmoplastic myxoid tumors of the pineal region (DMTPR) because they were extremely rare at our institution. Two patients had PTPR and no patient had DMTPR during the study period. Therefore, we included 43 patients in this study.
The following information was collected: age at diagnosis, sex, symptoms at presentation, tumor size, extent of resection, surgical approach, histopathological report, presence of hydrocephalus, leptomeningeal seeding (LMS), adjuvant therapy, disease progression/recurrence, and survival. The extent of resection was defined as follows: 1) gross total resection (GTR) as no residual lesion on postoperative MRI; 2) near total resection (NTR) as the removal of more than 90% of the tumor; 3) subtotal resection (STR) as the removal of 50%–90% of the tumor; 4) partial resection (PR) as the removal of less than 50% of the tumor; and 5) biopsy.
Histopathological diagnosis
The diagnoses were made according to distinct histopathological features of each grade of PPT.
PC is a well-differentiated pineal parenchymal neoplasm composed of 1) uniform cells forming large pineocytomatous rosettes, and/or 2) pleomorphic cells showing gangliocytic differentiation. Mitotic activity of PC is rare or absent, and Ki-67 proliferation index (PI) is less than 1% [11].
PB has histopathological features similar to those of embryonal tumors. It is a highly cellular, diffuse, and dense tumor composed of small blue cells. The shape of the hyperchromatic nuclei is irregular, and the nuclear-to-cytoplasmic ratio is high. Pineocytomatous rosettes are absent; however, Homer-Wright and Flexner-Wintersteiner rosettes may be seen. Necrosis is common and mitotic activity is high. The Ki-67 PI ranges from 20%–50% [2].
PPTID consists of diffuse sheets or large lobules of monomorphic round cells that appear more differentiated than those observed in PB. Mitotic activity is low to moderate, and Ki-67 PI ranges from 3%–20%. No criteria to satisfy the diagnosis of PB should be present. Although most PPTIDs fall under WHO grade 2, more aggressive cases may occur under WHO grade 3. There are no definite grading criteria to distinguish grades 2 and 3 [2]. Jouvet et al. [3] proposed grading criteria, grade 2 for PPT with less than 6 mitoses per 10 high-power fields (HPF) and positive immunostaining for neurofilament protein (NFP), and grade 3 for PPT with 6 or more mitoses or less than 6 mitoses with negative NFP. Our institution also followed to the criteria suggested by the study of Jouvet et al. [3].
Statistical analysis
Statistical analyses were performed using the R software version 4.2.1 (R Core Team, R Foundation for Statistical Computing, Vienna, Austria). Quantitative variables were presented as using median and interquartile range, whereas categorical variables were presented as frequencies and percentages. Due to the small sample size, we used the Kruskal-Wallis H test for quantitative variables and Fisher’s exact test for categorical variables to examine the differences in clinical variables between the three histological subgroups. Overall survival (OS) was defined as the time from the date of diagnosis till death. Progression-free survival (PFS) was defined as the time from the date of diagnosis to the date of the first recurrence. We used the Kaplan-Meier method for survival analysis. The log-rank test was used in the univariate analysis to determine the effects of prognostic factors on OS and PFS. Statistical significance was set at p<0.05.
RESULTS
Baseline characteristics
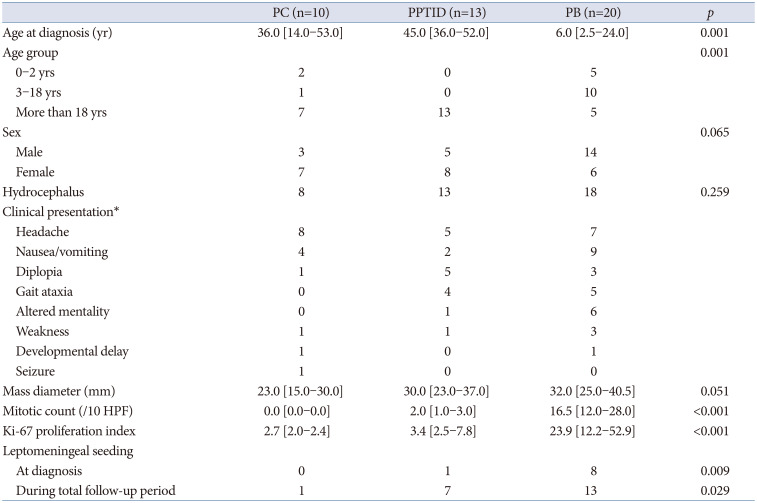
We identified 10 patients with PC, 13 with PPTID, and 20 with PB in our database (Table 1). All descriptions and analyses were based on initial diagnosis. The median age of patients with PC was 36 years. Children and adolescents accounted for 30% of PC cases. The median age of patients with PPTID was 45 years. None of the patients with PPTID younger than 19 years. The median age of patients with PB was 6 years. Most of the patients with PB were children and adolescents (75%). There was a female preponderance in the PC and PPTID groups. In contrast, approximately 70% of patients with PB were males. Most of the patients had hydrocephalus. Patients presented with various symptoms including headache, nausea/vomiting, diplopia, and gait ataxia, which are mainly related to hydrocephalus. The median diameter of the masses increased with increasing tumor grade. Mitotic count and Ki-67 PI also showed clear differences according to tumor histology. One patient with PPTID and 8 patients with PB had LMS at the time of diagnosis. One patient with PC developed LMS after malignant transformation of the disease to PPTID. During the follow-up, 53.8% of patients with PPTID and 65.0% of patients with PB experienced LMS.
Treatment
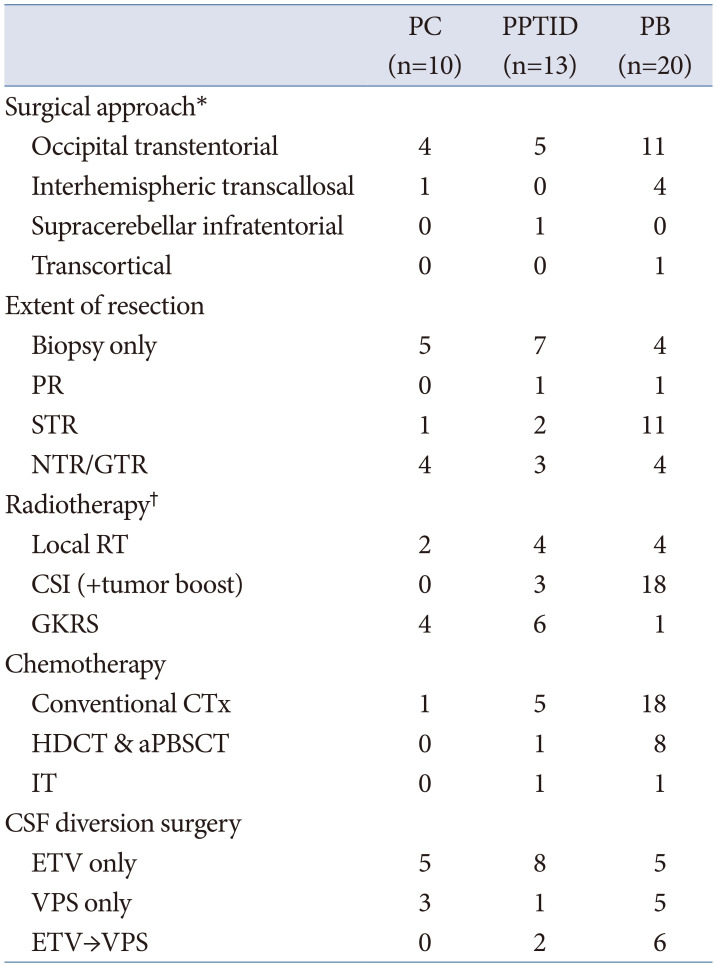
Table 2 describes the surgical and adjuvant treatment of PPT. The most preferred surgical procedure was the occipital transtentorial approach, followed by the interhemispheric transcallosal approach. Half of patients (50%) with PC and 76.9% of patients with PPTID underwent surgical biopsy simultaneously with an endoscopic third ventriculostomy (ETV). Four patients with PB (three of whom were adults) had endoscopic biopsy only and received adjuvant treatment. Five patients with PC, 6 with PPTID, and 16 with PB underwent surgical resection. Two patients with PC, 3 with PPTID, and 9 with PB initially underwent biopsy to confirm the diagnosis, and subsequently underwent craniotomy for the removal of the residual mass. GTR and NTR were achieved in 4 patients with PC, 3 with PPTID, and 4 with PB.
Six patients with PC, 11 with PPTID, and all patients with PB received radiotherapy (RT). Gamma knife radiosurgery (GKRS) was commonly used for PC (40%) and PPTID (46.2%). Craniospinal irradiation (CSI) was predominantly used (90%) in patients with PB. Among 18 patients who received CSI, 15 received CSI as an upfront adjuvant treatment. Two infants underwent delayed CSI after 36 months of age. A 74-year man received GKRS without biopsy at another hospital with an imaging diagnosis of PC. The size of the primary tumor initially decreased. After 2 years, he visited our institution with recurrent tumor accompanied by LMS, which was surgically diagnosed with PB. Subsequently, the patient underwent conventional chemotherapy (CTx) and CSI. Two patients with PB did not undergo CSI. CSI was delayed for a 1-year boy; however, his disease progressed rapidly. He had only palliative spinal RT and died. A 41-year woman was initially diagnosed with grade 3 PPTID. The tumor almost shrank completely after local RT. Ten years later, she presented with LMS of the cervical spine, diagnosed as PB. The initial diagnosis was reviewed by our pathologists and revised to PB. She has not begun adjuvant RT or CTx for PB.
Only one patient with PC received CTx. The patient experienced a malignant transformation of the disease to PPTID. She underwent CTx as adjuvant treatment after diagnosis with PPTID. Five patients with PPTID received CTx. A patient with grade 3 PPTID underwent conventional CTx, and intrathecal chemotherapy (IT) after LMS. Another patient with grade 3 PPTID underwent induction CTx and high dose chemotherapy (HDCT) with autologous blood stem cell transplantation (aPBSCT) after malignant transformation of the disease to PB. Of 18 patients with PB received CTx, 8 pediatric patients underwent HDCT with aPBSCT and one adult patient underwent IT. All patients who underwent HDCT or IT received upfront conventional CTx. Two patients with PB did not undergo CTx. A 2-year girl could not receive CTx as first-line adjuvant therapy due to shunt-related problem and her poor general condition. CSI was attempted, but it was discontinued due to leukopenia. Her guardian voluntarily rejected all treatments.
Of the 43 patients with PPT, 90.7% (39/43) had obstructive hydrocephalus. Among the 39 patients with hydrocephalus, 89.7% (35/39) underwent cerebrospinal fluid (CSF) diversion surgery, with 74.3% (26/35) of them receiving ETV with simultaneous tumor biopsy first. Nine patients who initially underwent stereotactic biopsy or craniotomy underwent shunt procedures. The effect of ETV was maintained in all patients with PC (5/5) and 80% of patients with PPTID (8/10). When there was LMS at diagnosis or when craniotomy was performed consecutively after endoscopic biopsy, the effect of ETV did not persist, and shunt surgery was inevitably required. Compared to the other groups, the transition rate from ETV to shunt placement was higher in PB group (54.5%, 6/11). Among all 8 PB patients with LMS at diagnosis, 4 underwent shunt placement initially, 4 received ETV first, which were later replaced with shunt.
Survival outcome and prognostic predictors
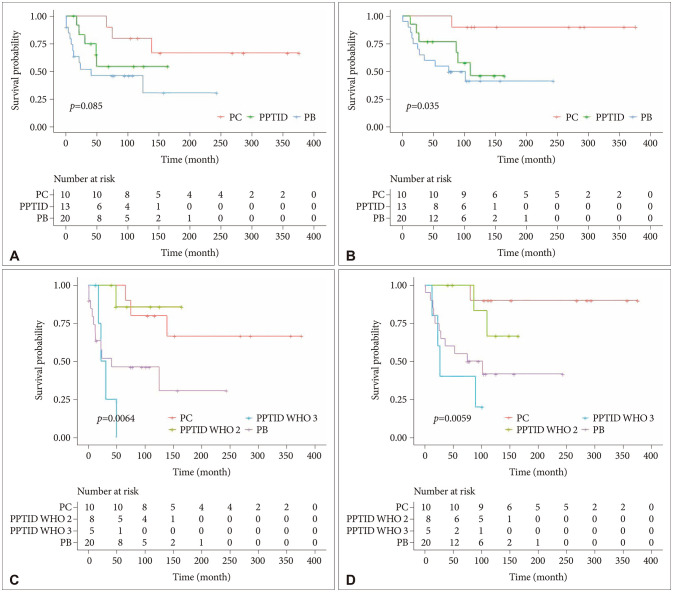
The median follow-up periods for PC, PPTID, and PB were 207, 90, and 75 months, respectively. When PPTID was analyzed at once without division by WHO grade, the prognosis was favorable in the order of PC, PPTID, and PB. The log-rank test indicated that a lower survival rate was associated with high-grade tumor histology (PFS, p=0.085; OS, p=0.035) (Fig. 1A and B). The 5-year PFS rates for PC, PPTID, and PB were 100%, 38.5%, and 40%, respectively. The 5-year OS rates for PC, PPTID, and PB were 100%, 61.5%, and 55%, respectively. Nonetheless, upon stratification of PPTID according to WHO grade, the prognostic outlook for grade 2 and 3 showed a clear disparity. Furthermore, grade 3 PPTID exhibited a worse prognosis than PB (PFS, p=0.0064; OS, p=0.0059) (Fig. 1C and D). The 5 year-PFS rates for grade 2 and grade 3 PPTID were 83.3% and 0%, respectively. The 5 year-OS rates for grade 2 and grade 3 PPTID were 100% and 40%, respectively.
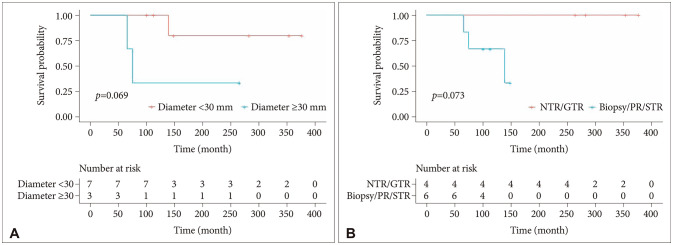
Since all patients with PC survived (except for one whose disease transformed to PPTID), we analyzed only the factors associated with recurrence in PC. Although the trend was not statistically significant, tumor diameter ≥30 mm (p=0.069) and a lesser extent of resection (NTR/GTR vs. biopsy/PR/STR, p=0.073) were associated with worse PFS in PC (Fig. 2). Four patients who had GTR did not experience recurrence. The tumor diameters ranged from 10 mm to 30 mm. A female patient with 36 mm-sized tumor underwent STR without adjuvant RT. Six years later, she received GKRS for grown residual tumor. Four patients underwent biopsy with adjuvant RT, including focal RT or GKRS. The tumor sizes of two patients who experienced recurrence were 26 mm and 34 mm, and those of other two patients without recurrence were 20 mm and 26 mm. A male infant who received biopsy without adjuvant treatment survived. The tumor size at diagnosis was 10 mm, and stabilized without increasing in size.
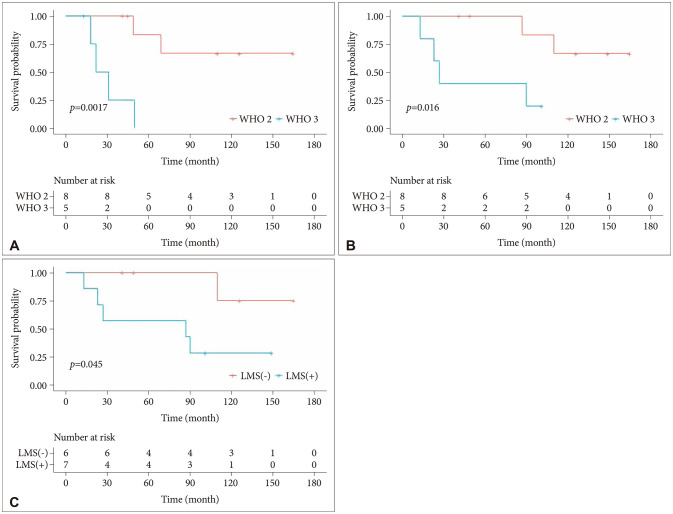
PFS (p=0.0017) and OS (p=0.016) were worse in patients with high WHO-grade of PPTID (Fig. 3A and B). Two of 8 patients (25%) with grade 2 tumors experienced recurrence. They initially did not have LMS and received endoscopic biopsy with adjuvant GKRS. Two patients (25%) with grade 2 tumors died. Four of 5 patients (80%) with grade 3 tumors experienced recurrence as LMS. All five patients with grade 3 tumors received upfront RT—local RT for 2, GKRS for 2, and CSI for 1—regardless of the extent of resection (including two GTR). A patient had an initial LMS and expired due to rapid deterioration of the general condition within 13 months of follow-up, despite adjuvant CSI. Four patients (80%) with grade 3 tumors deceased. LMS was a prognostic factor for worse OS (p=0.045) (Fig. 3C). Of the 13 patients with PPTID, only one had LMS at diagnosis; therefore, LMS could not be analyzed as a predictor of recurrence. One of 6 patients (16.7%) who did not suffer from LMS during the entire period of their illness died, while 5 of 7 patients (71.4%) who experienced LMS died.
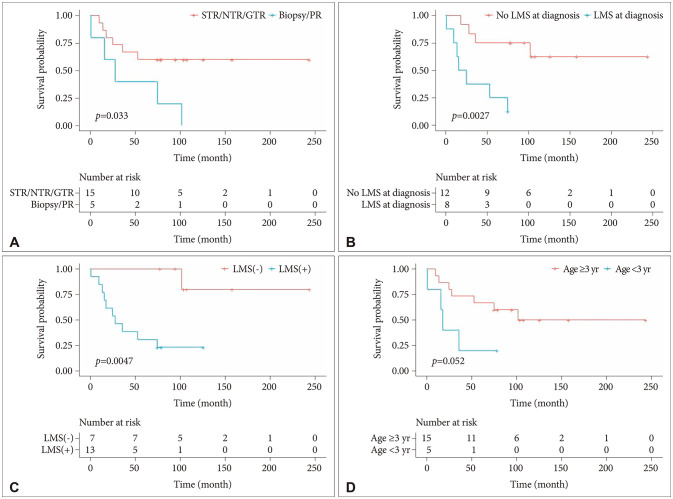
There were no prognostic factors for PB recurrence in any of the analyzable factors, such as age, sex, extent of resection, or LMS at diagnosis. A lesser extent of resection (biopsy/PR vs. STR/NTR/GTR, p=0.033) was associated with worse OS in patients with PB (Fig. 4A). All five patients who had biopsy or PR died, while 6 out of 15 patients (40%) who underwent more than PR expired due to their disease. A patient with PB died of secondary acute myeloid leukemia, which developed 5 years after the diagnosis of PB, although the primary lesion was cured after GTR without recurrence. LMS at diagnosis (p=0.0027) and LMS during the entire period of disease (p=0.0047) were poor prognostic factors for OS in patients with PB (Fig. 4B and C). Age <3 years at diagnosis showed the trend toward worse OS in all patients with PB (p=0.052) (Fig. 4D) and in the pediatric (0–18 years) subgroups (p=0.12). Among the patients receiving CTx, there was a better OS (p=0.17) with HDCT, although the trend failed to reach statistical significance.
Malignant transformation: three cases
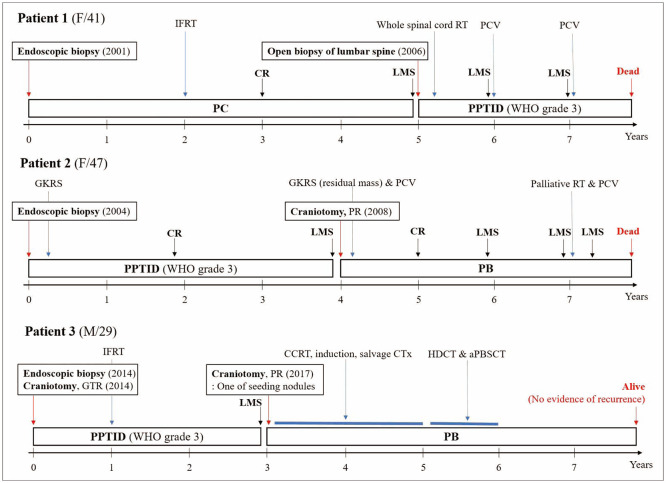
Three patients experienced malignant transformations (Fig. 5).
Patient 1 was a 41-year-old woman diagnosed with PC by endoscopic biopsy. She received 54 Gy of local RT 2 years after diagnosis. The tumor resolved on follow-up MRI. Three years later, LMS lesions at the lumbar spinal cord were confirmed. Grade 3 PPTID was diagnosed using open biopsy of the lumbar spine. She underwent whole-spinal RT and CTx with procarbazine, lomustine (CCNU), and vincristine (PCV). However, LMS aggravated, and she died 2 years after LMS identification.
Patient 2 was a 47-year-old woman diagnosed with PPTID via endoscopic biopsy. The mitotic count was 0 per 10 HPF, and the Ki-67 PI was 3%. Immunostaining of NFP was negative. Therefore, the tumor was classified as WHO grade 3 despite its low mitotic count and Ki-67 PI. This patient was reported by our institution in 2009 as a rare case of malignant transformation [12]. She underwent adjuvant GKRS (volume 12.4 cm3 with 13 Gy at 50%). The tumor disappeared on subsequent brain MRI. Four years later, she presented with dizziness and gait disturbance. Brain MRI revealed a 4 cm mass. She underwent craniotomy, and the mass was partially removed. The tumor was diagnosed as PB. The mitotic count was 15 per 10 HPF, and the Ki-67 PI was 19%. She received GKRS (volume 8.6 cm3 with 16 Gy at 50%) and PCV CTx. The brain lesion resolved completely on MRI 2 years after GKRS. However, other lesions at right internal auditory canal (IAC) and diffuse spinal LMS were found within a year. CSI could not be performed due to leucopenia. Despite palliative local RT (T12-S3 19.8 Gy and right IAC 25 Gy) and PCV retreatment, LMS worsened, and she deceased.
Patient 3 was a 29-year-old man diagnosed with grade 3 PPTID via endoscopic biopsy and consecutive craniotomy. The mitotic count was 12 per 10 HPF, and the Ki-67 PI was 8.91%. Increased cellularity and nuclear pleomorphism were observed; however, microvascular proliferation or necrosis was not observed. He received 61.2 Gy of local RT. A tiny solitary enhancing lesion remained on follow-up MRI. Three years later, the size of the enhancing lesion increased, and other intracranial LMS lesions were found. Craniotomy and biopsy were performed and the tumor was diagnosed as PB. The mitotic count was 60 per 10 HPF, and the Ki-67 PI was 30%. He underwent concurrent chemoradiotherapy (CCRT) including 23.4 Gy of CSI and 30.6 Gy of tumor boost with carboplatin and vincristine, followed by six alternative cycles of induction CTx. Induction CTx consisted of two protocols containing four anticancer drugs. One contained cisplatin, cyclophosphamide, etoposide, and vincristine, while the other contained carboplatin and ifosfamide instead of cisplatin and cyclophosphamide. After the treatment, the residual mass increased. Our oncologists changed CTx regimen to “8 in 1” including vincristine, CCNU, procarbazine, hydroxyurea, cisplatin, Ara-C, cyclophosphamide, and solumedrol. After 10 cycles of this salvage CTx, he underwent HDCT and aPBSCT including busulfan, melphalan, and thiopeta. Four years after HDCT, he is alive without recurrence. Brain and spinal MRI have been closely followed since the diagnosis of PB. He is the only survivor with grade 3 PPTID, even his disease transformed to PB.
Diagnosis assisted by molecular classification
Since 2018, our institution has used a next-generation sequencing (NGS) panel to diagnose brain tumors. Two patients were assisted by a novel molecular classification to revise or confirm their diagnosis. Aforementioned female patient initially diagnosed with grade 3 PPTID underwent surgery for cervical LMS. The diagnosis of LMS was PB because DICER1 mutation (c.1045_1054delGACACTTTCC) and partial deletion of chromosome 14q were confirmed in the NGS panel. The original diagnosis was reviewed and corrected to PB. Considering DICER1 mutation, loss of chromosome 14, and older age, this patient may belong to the PB-miRNA2 group according to recent consensus study [13]. Another male patient with grade 2 PPTID underwent surgical resection of lumbar LMS. KBTBD4 insertion (c.882_887dupCCCACG) was detected in the tumor NGS panel. The LMS lesion was also a grade 2 tumor. PPTID is characterized by recurrent KBTBD4 small in-frame insertions and the absence of DICER1 mutation or DROSHA homozygous deletion, which are typical molecular characteristics of PB [14].
DISCUSSION
The present study indicates that the prognosis of PPT obviously varies depending on the histopathological grade. In particular, grade 3 PPTID revealed a distinct clinical course compared to grade 2 tumors. Cases of rare malignant transformations were also observed.
PPTs commonly exhibit characteristic “exploded” calcification from the pineal gland toward the periphery and display low to moderate signal intensity on T1-weighted images and intermediate to high signal intensity on T2-weighted images, accompanied by notable contrast enhancement on MRI. PC appears to be an enlarged pineal gland and is well-circumscribed. These lesions have rare hemorrhage, less cellularity, and greater diffusivity than PB. Considering malignant nature of PB, indicators such as internal hemorrhage, necrosis, infiltration into adjacent structures, the presence of LMS, and diffusion restriction could be suggestive of PB. There are no distinct imaging features that can differentiated PPTID from PC or PB [15]. In this study, given the challenges of quantifying these imaging findings, only the tumor size and presence of LMS were addressed. Although PC and PPTID mainly affect adults and PB is more common in children, age and imaging findings are insufficient for a reliable diagnosis prediction of PPT.
PC is known to be controlled with appropriate surgical resection and adjuvant RT, with a long-life expectancy. Literature reported nearly 100% of 5-year PFS rate and above 85% of 5-year OS rate [161718]. According to Clark et al. [1920], the group that underwent resection demonstrated a better PFS than the group that received adjuvant therapy after biopsy. The GTR group benefited in both PFS and OS compared to the group that underwent STR with adjuvant RT. There was no significant difference in the effect of adding RT to STR on PFS and OS. We could not stratify patients with PC according to treatment modality because of the limited number of subjects. In the present study, the 5-year PFS rate was 100%. The GTR group showed better PFS than the group who had less than GTR with or without adjuvant RT. PFS ranged from 66 to 139 months in 3 patients who had tumor recurrence. If complete resection of PC is not achievable owing to surgical challenges, adjuvant RT is necessary, and a long-term follow-up of more than 10 years is required considering recurrence. Our analysis revealed that tumor diameter (based on 30 mm) was another predictive factor for recurrence. Tumor size may influence the choice of treatment and outcomes. The smaller the tumor, the easier the surgical resection and the better the result of adjuvant RT.
PPTID has an intermediate level of neoplastic behavior and treatment response compared to PC and PB [21]. Although PPTID is assigned to WHO grades 2 and 3, the definite histological criteria for WHO grading remain undefined. According to Jouvet et al. [3], grade 3 tumors are associated with aggressive behavior and poor outcomes compared with grade 2 tumors. In the present study, there was a marked difference in PFS and OS between grade 2 and 3, as previously reported [12223]. Patients with grade 3 showed worse prognosis than those with grade 4 PB. Our results suggest that certain subgroup of PPTID exhibit aggressive behaviors. The inclusion of 2 patients with malignant transformations in the grade 3 PPTID group in our analysis might make the prognosis of this group look worse. Several studies questioned the criteria of Jouvet et al. [3], and they set the grading with their own criteria using mitotic count and Ki-67 PI [72425]. Various prognostic factors including the extent of resection, age, and sex have been suggested in previous studies [1162326]. We could not confirm statistical differences in the outcomes according to factors other than tumor grade and LMS. There is no universally accepted treatment protocol for PPTID. STR is typically followed by adjuvant RT [242728]. Maximal resection or STR with adjuvant local RT may be suitable for grade 2 PPTID. Various platinum-based CTx regimens including PCV, have been used for grade 3 PPTID with LMS; however, specific indications and protocols are yet to be standardized [2229]. Yi et al. [30] reported on a case of grade 3 PPTID in which remission was achieved through PCV CTx following partial tumor removal and local RT. In this study, local RT was the primary adjuvant treatment for grade 3 PPTID. CSI or CTx was performed in some patients after LMS was identified; however, poor outcomes could not be avoided. Preemptive treatment, including CSI or CTx for LMS, should be considered for grade 3 PPTID.
PB is a malignant embryonal tumor with poor prognosis despite the implementation of radical surgical resection and multimodal adjuvant therapy. Age is an important factor affecting PB outcomes. A recently proposed molecular classification of PB supports differences in the biological behavior of this tumor with age [1331]. Hansford et al. [32] reported that the 5-year OS rate was 67.3% in children aged >3 years, and the 5-year OS rate was 16.2% in those aged <3 years. Despite intensive treatment, the survival outcomes of younger patients with PB were poor [333435]. Our data showed a trend similar to that reported in the literature, with a 5-year OS rate of 60% in children aged >3 years and 20% in children aged <3 years. LMS at diagnosis was a poor prognostic factor for OS regardless of age and treatment modality in the present study, as in previous studies [353637]. Several studies have reported that GTR is correlated with a better prognosis [43638]. However, there were studies that the extent of resection was not associated with survival outcome [3539]. Our data revealed that less than STR was associated with worse OS. Although controversial, it remains an important starting point for treatment for achieving maximal safe resection. Most patients with PB underwent adjuvant RT and CTx. Therefore, it was difficult to determine whether adjuvant treatment administered or not affected the prognosis. A differentiated treatment strategy is selected for children, especially those younger than 3 years, because of the neurotoxicity of CSI and aggressive features of the tumor at this age [35]. Despite the rare occurrence of PB, formulated treatment protocols are used based on the results of multiple clinical trials conducted on pediatric malignant brain tumors, such as medulloblastoma [323435]. Although there are specific differences in the protocols between countries and institutions, the common fundamental concept is to delay CSI in infants younger than 3 years of age and administer intensive chemotherapy during this period. Although there have been changes over time, the recent protocol applied to treat patients with PB younger than 3 years in our institution consists of maximal surgical resection, induction CTx with IT, HDCT/aPBSCT, and CSI at 3 years of age if LMS was identified at diagnosis [40]. In the protocol for patients with PB aged >3 years, CCRT with a reduced dose of CSI and boost are administered after surgery, and induction CTx considering HDCT/aPBSCT is performed. Gururangan et al. [41] reported that HDCT in addition to RT is an effective treatment for patients with newly diagnosed PB. A trend that HDCT/aPBSCT being associated with better OS was observed in the present study. However, transplantation-related complications are devastating; therefore, their occurrence must be carefully monitored and the adverse events have to be managed appropriately. Adult patients with PB have considerably less aggressive clinical course than in the pediatric patients [42]. In the present study, the 5-year OS rate in adult patients with PB was 80%, although the final survival rate was 40%. Adult patients in our data were managed differently compared to pediatric patients. Surgery followed by CSI and conventional CTx such as PCV regimen were the main treatment modalities, and HDCT/aPBSCT was not considered generally.
Pineal tumors frequently exhibit obstructive hydrocephalus early in the course of the disease because of the proximity of the mass to the cerebral aqueduct [43]. ETV is preferred to ventricular shunt because it provides the opportunity to conduct a biopsy of a bulging tumor in the posterior portion of the third ventricle, in addition to relieving hydrocephalus. Furthermore, a ventricular shunt carries the risk of tumor dissemination [16]. Approximately 15% of patients who underwent ETV may require ventricular shunt during follow-up. There were possible causes of ETV failure. The stoma can be occluded by the intraventricular tumor debris. Craniotomy following endoscopic biopsy may transform non-communicating hydrocephalus into absorptive type because of the release of proteins and blood into the CSF. LMS is another potential cause of ETV failure [44]. The relatively high rate of transition to shunt placement in patients with PB in the present study may be attributed to these reasons.
Endoscopic biopsy obtains a limited sample of tumor. Small samples may lead to misdiagnosis because they are not representative of the entire tumor, particularly in cases with mixed histology [4445]. In the present study, an 11-year-old boy was diagnosed with PC using endoscopic biopsy; however, the diagnosis was changed to PB after consecutive craniotomy. Our pathologists reviewed the specimen of the previous biopsy, and they concluded that the diagnosis of PC was appropriate for the specimen. Perhaps the tumor had some admixture of mainly PB and some tissues with characteristics of PC.
Malignant transformation of PPT has been reported in several studies [12294647]. The progression-free period ranged from 3 to 10 years. Most of the cases were transformed from PC to PPTID. We reported two cases from PPTID to PB. Whether the diagnosis is a true malignant transformation or a mixed pathology is debatable if the initial diagnosis was obtained by biopsy rather than resection. In the case of patient 1 who experienced malignant transformation in the present study, no detailed information other than the diagnosis was found in the pathological report when PC was diagnosed by endoscopic biopsy. In the case report of patient 2, specific justifications for establishing PPTID as a diagnosis in the first biopsy were provided [12]. When patient 3 was initially diagnosed and relapsed, each diagnosis was confirmed by open craniotomy. PPT can present as a mixture or a continuous spectrum of low-grade and high-grade tumor. Over the past few years, the biology and inter-tumor heterogeneity underlying PPT have been clarified through multi-omic research [131431]. Recent molecular classification of PPTID and PB provide accurate diagnosis through biopsy, help confirm the diagnosis of recurrent lesions, and enable comprehensive prognostic prediction.
The present study had some limitations. This study was constrained by a small sample size and the inherent biases of a retrospective design. Recently, several studies have pooled multicenter data or used nationwide registry such as the Surveillance, Epidemiology and End Results (SEER) or the National Cancer Database (NCDB) [417232632353942484950]. Despite having less statistical power than larger-scale research, our study is meaningful for evaluating our past clinical practice, comparing it to other studies to identify the strengths and weaknesses of our institution, and finding areas for future improvement in PPT patient care. A prospective multicenter trial is needed in the future. Because of the rarity of PPTs, we pooled the data from adult and pediatric department. The treatment strategies of each department were not uniform. Establishing standard treatment protocol is essential. Since the application of tumor gene panel has not been long, few cases reflected novel molecular classifications in diagnosis. If the molecular classification of PB and PPTID is employed, more precise diagnosis and prognostication will be feasible.
Four WHO grades had different impacts on the prognosis. In particular, grade 3 PPTID shows aggressive behavior and dismal prognosis. Maximal surgical resection is critical; if it is unavailable, appropriate adjuvant treatment should be administered timely. Performing imaging evaluation, especially spinal surveillance, at close intervals and preemptively applying adjuvant treatment in preparation for LMS is important in the management of grade 3 PPTID. Considering the possibility of malignant transformation, long-term attentive follow-up is required.
 XML Download
XML Download