

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Post-stroke spasticity (PSS) and post-stroke spastic movement disorder (PS-SMD) are common conditions following stroke that include damage of the sensori-motor networks in the central nervous system (CNS) [1]. PSS is characterized by involuntary activation of skeletal muscles resulting in phasic and/or tonic muscle activity during rest and voluntary movement resulting in the PS-SMD in involved body-parts [2]. PSS is the result of the so-called positive signs of the upper motor neuron syndrome (UMNS). These positive signs of the UMNS are (a) spontaneous or triggered (stretch- or touch-induced) clonus activity or spasms during rest or movement; (b) spontaneous antagonistic co-activation during rest called spastic dystonia; (c) involuntary spastic co-contraction of the antagonist while voluntary activation of the agonist; (d) velocity-dependent increase in muscle tone during rest; (e) increased tendon reflexes; and (f) Babinski sign and other pathological reflexes during rest or triggered by touch or movement [2,3].
Most of the prevalent data of PSS originates from studies that only used the criterion of velocity-dependent increase in muscle tone as a clinical marker of PSS [1,2]. This prevalence is high in the first year after first ever stroke, affecting up to 43% of stroke survivors [1,2,4]. PSS, resulting PS-SMD and complications can be a major contributor to stroke-related disability, low quality of life, and reduced social and professional participation [2]. PSS or PS-SMD that is perceived by the affected individual or care giver as hindering body functions, activities, and/or participation is defined as disabling PSS or disabling PS-SMD [2,4]. Published prevalent data of a severe spasticity (defined as increased muscle tone equal or more than 2 of the modified Ashworth scale [MAS]) and a disabling PSS (defined as spasticity that does need treatment) showed that up to 13%–16% of stroke survivors suffer from this complication after stroke [2,4,5].
The symptom velocity-dependent increase in muscle tone, characterized by resistance to passive stretch of the affected skeletal muscles, is still the key qualitative symptom that leads to the diagnosis and allows to characterize the topical distribution of the PSS over the body regions [2]. The documentation of the muscle tone with the Ashworth scale (AS), MAS, or Tardieu scale (TS) as a clinical quantification of the PSS is well established in most of the interventional and pivotal studies [2]. For a state-of-the-art-documentation and a precise communication of the topical distribution of the PSS the classification of the distribution as focal, multi-focal, segmental, multi-segmental, or generalized PSS is recommended [3,6,7]. For a standardized calculation of the severity of e.g., hemispasticity, para-, or tetraspasticity for calculation of sum scores the REsistance to PAssive Stretch scale (REPAS) with defined test positions and set of well standardized passive stretchings using the AS is recommended [8].
PS-SMD is associated with negative features of the UMNS namely muscle weakness, fatigue and fatigability creating dexterity problems with slowing and less forceful or even no voluntary movements in the affected body region. Therefore, the term spastic paresis (SP) is also used to describe the clinical picture on an impairment level in PS-SMD since it combines PSS (involuntary muscle activity) with muscle weakness, which are commonly present in the UMNS [2,9].
The UMNS with a PS-SMD consists of neuronal changes and progressive muscle and soft tissue changes. These changes shorten the involved structures and lead to a progressively reduced range of motion (ROM). This aspect of the UMNS is called the non-neuronal component, which led to the establishment of the term deforming spastic paresis (DSP). In the 2005 definition of Pandyan et al. [9], this non-neuronal component and the negative sign paresis of the UMNS are not part of the SPASM definion of spasticity.
Another severe complication resulting from a PS-SMD is the syndrome of spasticity-associated pain, which could be diagnosed by eliciting stretch-induced nociceptive pain in spastic muscles in affected body regions [2,10]. The pathophysiology is not totally understood, but the hypothesis is that it correlates with the quantity of some positive features of the UMNS (especially spastic dystonia) and also with malpositioning, sensory loss, and neglect syndrome that affect the involved limbs. Large randomized controlled trials (RCTs), cohort studies, and pooled data analysis of stroke survivors in the chronic phase showed that up to 64% of patients with PS-SMD had pain in the paretic limbs to some degree [10]. Both, randomized controlled studies and pooled RCTs [10] demonstrated a significant reduction of spasticity-associated pain if muscles are treated with botulinum toxin A (BoNT-A). This applies for all a focal, multi-focal, and segmental BoNT treatment regimens.
Currently, there is still no causal treatment for PSS and the resulting PS-SMD. However, various symptomatic therapeutic approaches have been introduced and discussed for managing PS-SMD [2]. In this article we provide information concerning the pathophysiology of PSS and its prediction, as well as the clinical assessment, management strategy including goal setting and multimodal treatment options with a focus on evidence-based methods and BoNT-A treatment.
PATHOPHYSIOLOGY OF PS-SMD
For the first time, an international group of specialists in neurology, neurorehabilitation, and restaurative neurology defined the different relevant pathophysiological phases following first-ever stroke [11]. According to the panel, there are four main phases after acute ischemic or hemorrhagic lesion of the cortical and subcortical brain tissue. These phases show specific histopathological and/or pathophysiological correlates: The Hyperacute Phase (within the first hours to 24 hours), the Acute Phase (first day to a week), the Subacute Phase (after first week, up to 6 months), and the Chronic Phase (after 6 months) [1,11,12].
In the hyperacute and acute phase necrosis of the brain tissue happens with local inflammatory and secondary neuro-degenerative processes at and around the lesion site. In the first hours after stroke cortical and subcortical sensori-motor networks structures show acute loss of function in involved movement segments on the contralateral body side with acute flaccid paralysis and/or sensory loss. In this phase the limb is dependent on passive positioning, as there is a high chance of no voluntary control of the position of the limb. The loss of sensation results therefore in a high risk of secondary damage due to malpositioning [11-13].
With a latency of days to weeks neuroplasticity starts with re-connection of residual neurons and rewiring of new connetions of the lesioned area to new neuron pools in the sensori-motor network nearby. These complex and often overlapping processes happen with variable latency (days to weeks) in the late acute and ealy post-acute phase [11,12]. From epidemiological studies of large cohorts of stroke survivors, it is known that about 20% develop a SP [1]. The emerging regain of movements to a certain extent is paralleled by involuntary muscle activity, that is defined as spasticity [9,14]. In other words, the start of regaining of some voluntary muscle force and sensation could be concurrent altered by involuntary muscle activity, which is defined as spasticity or positive signs in the context of an UMNS [9,13,14].
The involved neuronal mechanisms resulting in an increased excitability of the sensori-motor spinal networks and promoting involuntary muscle activity are due to an imbalanced descending regulation of the spinal sensori-motor network. There is an imbalance between descending inhibitory dorsal reticulospinal tract (RST) and the excitatory medial RST and vestibulospinal tract and/or abnormal intraspinal processing of sensory input (enhanced sensitivity of Ia and IIa afferents, and reduced presynaptic inhibition on Ia-afferents, as well as facilitation of group Ib and II afferents) [13,15-17].
Additionally, there is also evidence of a shift of the chloride equilibrium potential in spinal cord motoneurons due to an altered function of the chloride extrusion mechanisms (KCC2) resulting in excitatory effects of GABA and glycine on motoneurons [18].
In this stage the affected limb develops to some extent the so-called spastic movement pattern that shows typical dysbalanced movements, joint positions with slowing in its performance compared to physiological movements on the less affected body side. By definition, if involuntary muscle activation is included in those typical movement patterns in a central paresis, this syndrome represents a PS-SMD [12].
Whether regaining of any sensori-motor function ends with either a residual loss of function or a regaining of most of the functions in the upper and lower limb in the late post-acute or in the chronic phase seems to be dependent of various factors. For sure, the lesion size in the sensori-motor cortical and subcortical network is important for both regaining any function [19] and for the development of spasticity/PSS [20,21]. The following clinical signs represent negative predictors for a good outcome and an high risk for the development of a severe PS-SMD: severe sensory loss, neglect or other relevant neuropsychological changes, paralysis or severe paresis without development of functional relevant muscle force (MRC below 3), severe PSS in multiple joints and adaptation to a severe PSS (MAS= or >than 2), development of a DSP with contractures and spasticity-associated pain [2]. The progressive so-called non-neuronal changes of muscles and soft tissue in the late post-acute and chronic phase (DSP) may lead to further functional impairments with progressive joint contractures, spasticity-associated pain and skin irritation or even skin lesions [22,23].
In summary, the Acute and Early Subacute Phase (first weeks to 3 months) is characterised by the start of reorganisation of the sensori-motor network. In this phase SP develops and involuntary muscle activity add the risk from paresis and may result in the development of complications from PS-SMD with development of contractures, spasticity-associated pain and DSP [1]. In the Late Subacute Phase (3–6 months) in most cases the development of PS-SMD is established and the sensori-motor system shows signs of (mal-) adaptation to involuntary muscle activity in up to 42% of stroke survivors [1]. In 13% this led to a syndrome of disabling PS-SMD [2,4]. Later on, in the Chronic Phase an established PS-SMD may further worsen with progressive immobility, joint contractures, stretch-sensitive spasticity-associated pain, and abnormal movement patterns [4,12].
The knowledge of the pathophysiology of the PS-SMD and its different time phases following a first-ever stroke is crucial for appropriate management strategies of an underlying UMNS with high risk of the development of a DSP [3,11,12,22]. However, the interconnection between the various facets of the neuronal and non-neuronal mechanisms of PS-SMD with involuntary muscle activity, defined as spasticity by the SPASM-group [9], on the one hand, and progressive muscle degeneration (loss of myofibrills) and muscle and soft tissue shortening and stiffening are not yet fully understood [22-24]. The time scales of mechanisms of degeneration and reorganization parallels to some extent the time scale of neuroplasticity of the sensori-motor nervous system and peripheral tissues in the subacute phase [17,23].
Several cohort studies could show that changes in muscles and peripheral soft tissue (changes in tissue matrix, storing of collagen, degeneration of muscle fibres) starts already within a few weeks after first ever stroke [11,12]. Unfortunately, it is not fully understood yet, whether there are specific risk or trigger factors causing those tissue changes or whether it is the natural course after a first ever stroke.
Because there is currently no causal treatment for PS-SMD and it seems to develop in the Acute and Early Subacute Phase following stroke, it is crucial to catch the symptoms early and identify potential predictors of a PS-SMD to promptly initiate an appropriate management consisting of both physical and medical treatment [2]. Given the high prevalence of stroke as well as of a disabling PS-SMD, it has been strongly recommended for any initiating efforts to detect and treat early symptoms of PS-SMD as early as possible [2,12]. This is to achieve effective symptomatic management and avoid complications and maladaption to PSS.
Early PSS detection and management may imply a better chance for a better outcome of patients, who passed a comprehensive person-centered neurological rehabilitation program following stroke. Timely and appropriate physical management and medical treatment, including BoNT-A treatment, seems to be crucial to prevent further disability from PSS to DSP in the context of a PS-SMD and may improve the outcome for individuals suffering from PS-SMD [2].
MANAGEMENT OF PS-SMD
The prediction of PS-SMD and goal setting
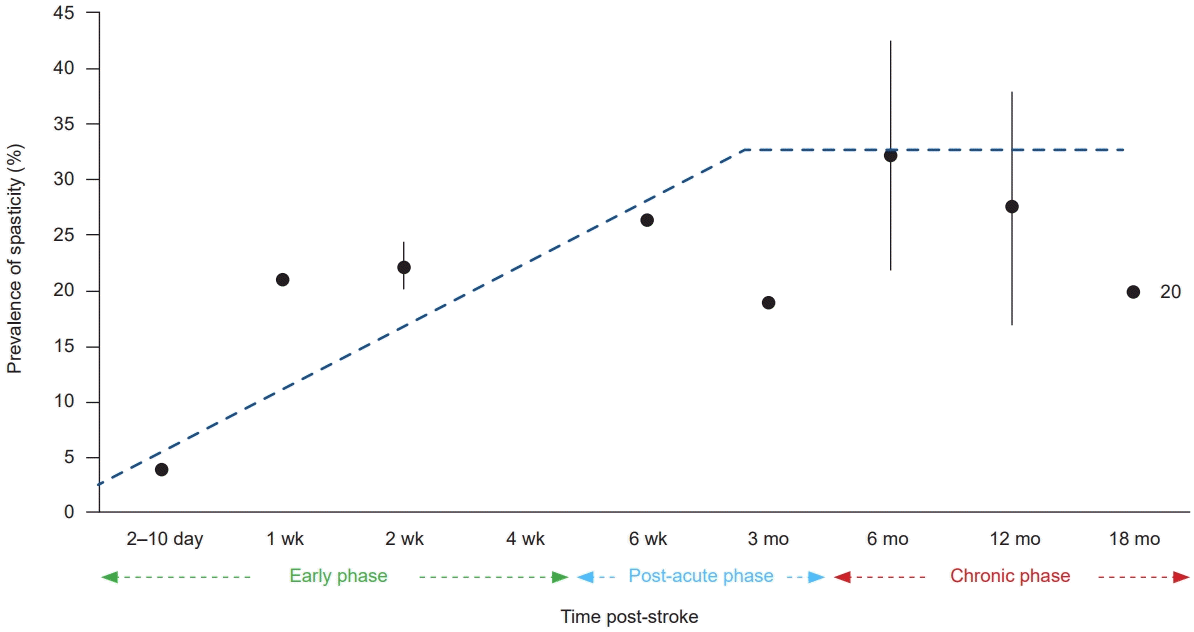
The PS-SMD typically emerges between days and three months after the first-ever stroke. The prevalence of a PS-SMD increases with time and reaches a plateau at about three months after the stroke (Fig. 1) [1,2].
Chronic PS-SMD is considered as a phenomenon of post-stroke maladaptation, associated with neuronal re-organization within the CNS during a critical period of three to six months after stroke [17]. This three to six-month period is characterized by complex processes involving immunologic responses that aim to limit the damage caused by stroke but can also lead to further damage to the affected tissue [2,17]. The window of neuroplastic re-organization might increasingly narrow over six months indicating the importance of early detection and intervention to avoid hardly reversible maladaptation to PS-SMD [1,2].
Prediction of PS-SMD
Many studies have identified clinical and brain-imaging data as predictors for early detection of patients at high risk of developing disabling PS-SMD (Table 1) [25-35].
Based on published predictors of PS-SMD a group of experts in the field published “The PSS Risk Classification System” to allow for prediction and identifying patients at risk to develop a PS-SMD within the acute and post-acute phases following a stroke [35,36]. This tool focuses on early identification of clinical and imaging risk factors and provides recommendations for managing PS-SMD.
Recent publications have identified additional red flags for predicting a PS-SMD. This includes high scores on the modified Ranking scale (mRS), the National Institute of Health Stroke Scale (NIHSS), and lower than 27 scores for mini-mental status examination (MMSE) within the first week after first ever stroke [37].
In addition to these clinical predictors, specific brain lesion localizations and volume were found to predict a PS-SMD, too. Lesions involving the basal ganglia, thalamus, insula, and white matter tracts (such as the internal capsule, corona radiata, external capsule, and superior longitudinal fasciculus) on cerebral magnetic resonance imaging scans taken in the first week after stroke have a high predictive value for PS-SMD when the lesion load affects the cortico-spinal tract. Larger lesion sizes in these areas also increase the risk for developing a PS-SMD [2,20,21].
Goal setting and attainment
Before starting with a rehabilitation program, a systematic examination of the patient with spastic movement disorder is necessary to identify the relevant problems on the levels of impairment, activities, and participation of the individual patient. This examination should include a detailed case history of the stroke with a detailed description of the evolution of the spastic movement disorder. If relevant, also other medical issues should be evaluated. This should be followed by a comprehensive neurological examination by the physician as well as a clinical evaluation by the members of the multiprofessional therapeutic team, specialized in the management of spastic movement disorder [2].
On the basis of this thorough evaluation realistic short- and long-term goals for the comprehensive person-centered rehabilitation program have to be established, always in collaboration with the patient/caregiver and members of the multiprofessional team (MPT). An expert physician in neurorehabilitation, specialized in the management of spastic movement disorder, should guide this important discussion on goals for the rehabilitation program. Patient-centered goal setting, aligned with the International Classification of Functioning, Disability and Health, can help to manage activity limitations or functional impairments [2,38].
Collaboration among patients, caregivers, clinicians, and the MPT is essential to define desired treatment outcomes for BoNT-A injections in a multimodal treatment program. The SMARTER matrix, which stands for Specific, Measurable, Agreed, Realistic, Time-bound, Evaluated, and Revised goals, can help in developing realistic individual goals. After agreement of realistic goals, the team formulates a PS-SMD-rehabilitation plan adapted on the patient’s individual needs and abilities. If the impairment positive symptoms of a UMNS result in a SMD with a focal, multi-focal or segmental distribution, the multiprofessional rehabilitation program should include a management with BoNT-A [2,39].
Goals selected together with the patient and/or caregiver can be categorized into two domains: (1) symptoms and impairment, such as pain, involuntary movements, and ROM/contracture prevention, and (2) activities and functions, including passive and active function, as well as mobility [2].
During the individualized rehabilitation program assessing goal attainment is a main component of clinical assessment and optimal management of PS-SMD. Goal attainment scaling (GAS) is a commonly used tool for evaluating treatment goals, where patient-specific SMARTER goals are tracked using a standardized scale. It encourages patient’s engagement in goal setting and has a positive impact on goal achievement, with evidence showing that involving patients in goal setting increases their likelihood of success [2,39].
The management of PS-SMD with BoNT-A
The efficacy of BoNT-A in the treatment of PS-SMD has been extensively studied. BoNT-A is considered as the treatment of choice for focal, multi-focal, and segmental PS-SMD, including spastic hypertonia, increased muscle tone, spastic dystonia, clonus, and spasms [2,3,5,6].
Numerous double-blind studies and systematic reviews have demonstrated improvements in reducing muscle tone and passive function in spastic limbs after stroke [2,3,12]. BoNT-A has been shown to reduce spasticity-associated pain, improve hygiene by increasing the passive range of joint movements, and reduce malpositioning of limbs caused by spastic dystonia and shortening of muscles due to spastic movement patterns [2,12,39]. It has also been proven to enhance active functional gains in certain subgroups of patients with PS-SMD who have difficulties with active movements due to simultaneously activated antagonists or increased muscle tone of antagonist muscles, such as reaching, gripping, or relieving movements with the hand or antagonistic ankle movements [40].
The onset of the effect of BoNT-A in PS-SMD typically occurs between 2 and 5 days after intramuscular injection, with maximal effect observed at 3–4 weeks. This clinically significant effect of BoNT-A lasts for 6–10 weeks, after which it gradually declines. The duration of the effect mainly depends on the injected dose of BoNT-A per muscle. As the uptake in the motor endplate of BoNT-A is an active process, induced contractions by passive stretch an/or electrical stimulation of the injected muscle after BoNT-A injection may help to increase and extend its effect size and duration. The overall effect of BoNT-A treatment typically lasts for about 3-6 months following injection [2,41].
Controlled studies have shown that early BoNT-A intervention within 3 months following stroke, can result in a longer duration of improvement of both spastic muscle tone and passive function while reducing the occurrence of complications like tendon shortening in long finger flexor tendons with a lower dosis of BoNT-A per injected muscle [2,12,42]. However, up to now the evidence does not indicate higher functional gains or effects on disability with earlier interventions with BoNT-A following stroke.
BoNT-A treatment in SMD has a good safety profile when used within the recommended therapeutic dose per muscle and treatment session and with proper injection techniques [2]. Common local adverse events include unwanted weakness of injected muscles, dysphagia (injection in cervical muscles), dry mouth and eyes, local pain, bleeding, or hematoma at the injection site. These side effects are reversible and not life-threatening. Systemic adverse events are rare and seemed to be less frequent if recommended doses are injected by use of guidance techniques directly into the target muscles in PS-SMD [2,41].
In the upper limbs, BoNT-A management of spastic movement disorder has shown dose-dependent effects in reducing spastic muscle tone, improving passive ROM and passive function, and reducing caregiver’s burden. However, the effects on active functions of the hand or arm are limited [2,39]. BoNT-A injections may improve active function in some patients with spasticity of the upper limb, especially when spastic co-contraction of antagonistic muscles is the relevant cause of hindering active finger or elbow from movement. In such cases the injection of BoNT-A in spastic agonists is recommended to combine with active training of motor command and force for antagonistic muscle groups thereby allowing for improvements also in active function, e.g., finger, wrist, or elbow extension [2,10,39].
In the lower limbs, BoNT-A injections can be considered for clinically relevant lower limb PS-SMD that does not respond to conventional physiotherapeutic treatment. BoNT-A injections were shown to reduce spastic muscle tone and to improve passive joint mobility and joint position in the ankle, knee, and hip. Pivotal studies in the lower limb of BoNT-A products showed significantly reduced pes equinus and pes equinovarus position with reduced muscle tone in the calf muscles and improved brace or orthosis tolerance. However, significant improvements in longitudinal gait parameters (gait speed and step length) have not been consistently observed [2,40,43].
BoNT-A treatment also showed reduced spasm frequency and clonus occurrence, as well as reduced stretch- or motion-related spasticity-associated nociceptive pain in both upper and lower limbs after stroke [2,3,6].
The technical guidance of BoNT-A injections, such as ultrasound or electrical stimulation guidance, improves the accuracy, safety, and efficacy of the injections compared to non-guided injections. Guided injections have been shown to be superior in terms of injection accuracy and avoidance of accidental vessel and nerve injury [2,41].
The appropriate dosing of BoNT-A is crucial for treatment effectiveness and to minimize adverse events. Dosing recommendations are currently based on limited studies and rely on pivotal studies that are documented in the product information, clinical experience of injectors, and expert consensus statements [2,3,41].
Recommended doses per injection site base on the assumption of a saturation of the motor endplates and led to the recommendation not to inject more than 50 units for onabotulinumtoxinA and incobotulinumtoxinA, and 125 units for abobotulinumtoxin A per injection site. Therefore, the number of injection sites per muscle depends on the maximal recommended dose per specific muscle. As a a rule of thumb the recommended doses vary for different muscle sizes, ranging from 15–25 units onabotulinumtoxinA and incobotulinumtoxinA, and 35–75 units for abobotulinumtoxin A for small limb muscles to 50–150 units onabotulinumtoxinA and incobotulinumtoxinA, and 125–500 units for abobotulinumtoxin A for large limb muscles [2,41].
As spastic muscle patterns of upper and lower limbs usually involve more than two or three muscles it seems to be important to calculate the total dose per injection session prior to the event and respect the maximum dose per injection session given in the product information and from expert consensus statements [2,41]. There are prospective studies that could show a superiority with respect to the benefit with higher dose, but for certain BoNT-A products, only [44]. More research is needed in this field to learn more about the safety margins of the different BoNT-A products.
Adjunctive therapies to botulinumtoxin injections
Evidence-based reviews report an enhanced effectiveness of adjuvant therapies to BoNT-A injections in spastic muscles in PS-SMD [2]. Such techniques include physiotherapy, modified constraint-induced movement therapy, electrical stimulation, casting, and dynamic splint treatment. Neuromuscular electrostimulation applied three days before and after BoNT-A therapy has shown positive effects on effect size and duration of BoNT-A injections. Other adjuvant therapies like stretching, taping, and robotic training may be used on an individual basis, but the published evidence on additional effects is limited [2].
Oral antispastic drugs have shown significant systemic side effects like sleepiness, drowsiness and general weakness and do not have sufficient evidence to support their superiority over local BoNT-A treatment in focal, multi-focal and segmental spasticity. But some studies have demonstrated effectiveness of systemic drugs like tizanidine or baclofen [2]. On the basis of clinical experience, in some patients the drug side effect ‘sleepiness’ may help for positive effect on lower limb spasms during night. Such timed drug therapy may be combinable with BoNT-A injections for treatment of a pes equinus or ankle clonus within the calf muscles.
A combination of diverse treatment modalities involving neurolysis (phenol, alcohol, or cryo-neurolysis) or even neurotomy with BoNT-A injections can be considered for PS-SMD that shows a multi-segmental or generalized topical distribution and does not respond adequately to recommended dose per session [2,45]. If the BoNT-A dose per session exceeds the limit documented in the product information or in consensus statements a combination of neurolysis of motor end branches or dominat motor nerves, like musculo-cutaneus or obturator nerve, are recommended in order to reduce the total dose of BoNT-A per session. However, the side effects and the long-term side effects of neurolysis should be taken into account [2,45].
Intrathecal baclofen (ITB) application can be considered for severe multi-segmental or generalized PS-SMD that does not respond to other interventions, e.g., systemic oral pharmacological antispastic treatment [2,3]. For ITB therapy was shown a superiority over conventional medical management with oral antispastics in terms of efficacy and pain control [46]. Surgical procedures, such as fasciotomy, tendon and muscle lengthening, and tendon transfer surgery, may be considered in chronic PS-SMD cases after exhausting of other reversible treatment options [2]. More research is needed to provide evidence for orthopedic surgery in the management of chronic PS-SMD and its combination with BoNT-A treatment.
BoNT-A therapy in the Acute and Early Subacute Phase following stroke
PS-SMD typically develops in the early post-acute phase of stroke, within the first three months [2,12]. Unfortunately, clinical detection and decision-making for appropriate management in many countries usually occur later, e.g., in the late sub-acute or even in the chronic phase [1,2]. In the late sub-acute phase degenerative changes in affected muscles and soft tissues already start and lead to more frequent complications like joint contractures, spasticity-associated pain syndromes, abnormal motion and limb patterns and even bony distortions can occure in the chronic phase - if the SMD is not adequately managed in the early sub-acute phase [2,13,17].
Studies have shown that early initiation of treatment after stroke could have beneficial effects on the reduction of spastic muscle tone without increased side effects [2,12,47]. Meta-analysis and systematic reviews indicate more favorable outcomes and better prognosis with early treatment compared to late treatment in the chronic phase of PS-SMD [2,12,42].
Based on the pathophysiological mechanisms leading to a SMD it has been considered to maintain or provide a normal sensory and proprioceptive input to the disrupted central sensorimotor network to prevent from adaptation or even mal-adaptation re-organization. BoNT-A injections in SMD developing muscles support to prevent the central network from overactive peripheral sensory input from intrinsic muscle fiber and tendon receptors (IA- and IIb-fibers). The pathologically enhanced tonic and phasic input on the spinal sensori-motor network via the proprioceptive system is believed to be one of the key factors for maintenance and augmentation of the spastic movement disorder [12,13,16,48]. It is hypothesized that this input to the spinal network from spastic muscles may play a significant role in the maladaptive processes in the CNS that leads to the chronic consequences from PS-SMD in UMNS [13,15-17,48].
It is suggested that the three-month transition phase following stroke is a period of enhanced neuroplasticity allows effective interventions. Such interventions include blocking afferences from spastic muscles thereby avoiding the development of complications like contractures and spasticity-associated pain syndromes. This is achievable by BoNT-A treatment in spastic muscles before the transition in the late post-acute phase. Therefore, this transition marks the cut-off for early vs. late BoNT treatment [12].
Based on the evidence that appropriate predictors are sufficient to detect patients at risk for developing a PS-SMD after a first ever stroke, it seems now possible to treat PS-SMD with BoNT-A already in the acute or late early subacute phase, i.e., within the first 3 months after stroke [2,12,42]. Treatment of PS-SMD with BoNT-A injections should be offert to those high risk patients to avoid complications from severe spasticity, e.g., contracture development. As well, involuntary muscle activity should be blocked early to avoid maladaptation to a disturbed feedback on the spinal network by increased Ia- and II-afferences from hyperactive muscle activity, e.g., from spastic dystonia. Spinal cord sensori-motor networks can be protected from an enhanced muscle/tendon proprioceptive input by early BoNT treatment. By doing this, the vicious circle can be interrupted and neurorehabilitation is facilitated. Insofar, in patients, who are at high risk for a disabling spasticity it is recommended to start a BoNT therapy early, when early signs like moderate increase in velocity dependend increase in muscle tone (MAS >1+) is present in two joints of the paretic limbs [35]. Goal-oriented management of PS-SMD, including BoNT-A therapy within a few weeks to three months after stroke onset, has shown to prevent or reduce the development of severe or disabling SMD and its complications like finger contractures [2,12]. Considering positive outcomes associated with early BoNT-A therapy, it is strongly recommended to initiate treatments including BoNT-A therapy in the acute to early subacute phase after stroke in patients with existing PS-SMD or at high risk of developing severe PS-SMD, to prevent or reduce post-stroke disability and improve rehabilitation outcome [2,12,42]. This is especially true for BoNT-A injections within 3 months following stroke in patients with beginning PS-SMD or at high risk to develop severe or disabling spasticity as controlled studies with early BoNT-A interventions showed longer endurance of BoNT-A effect with lower dose per muscle compared with dosis used in chronic spasticity and reduction in the probability to develop complications like contractures and spasticity-associated pain in the chronic phase after stroke [2,12,42].
In the chronic phase of focal, multi-focal, and segmental PS-SMD BoNT-A injections should primarily target spastic muscles that are either identified to create disability or muscles that already are involved in complications It should be underlined that therapeutic interventions for severe or disabling PS-SMD should be embedded in a multi-professional team approach that involves a comprehensive patient-centered multi-modal therapy program. This includes e.g., occupational and physical therapy, physical treatments (casting), electrical stimulation, and BoNT-A injections to the targeted spastic muscles [2,3].
Regardless of the fact that the majority of BoNT-A treament for PS-SMD is done in the chronic-phase after stroke - where spasticity and muscle deformities are already established, it should be noted that there is no restriction for the timing of BoNT-A treatment for any region of the upper and lower limbs based on product licenses.
Degenerative changes in affected spastic muscles
The long-term repeated or chronic use of BoNT-A injections for spasticity management has raised concerns about persistent muscle atrophy and degenerative changes in the affected and treated muscles [49]. Affected spastic muscles naturally undergo degenerative changes, including muscle fiber atrophy, fibrosis, and altered architecture. Based on that information the term “spastic myopathy” has been coined by Gracies [22]. As well repeated BoNT-A injections might cause changes in the muscle and might exacerbate these natural changes through muscle atrophy, fibrosis, and altered muscle fiber composition [49].
Prolonged dis-use from non-use in SP can cause muscle atrophy as well as atrophy from temporary neuromuscular endplate block of BoNT-A, leading to decreased protein synthesis and increased degradation. Chronic denervation resulting from BoNT-A injections may trigger fibrotic changes in spastic muscles via gene expression changes, reduced trophic support, and inflammation, leading to the deposition of collagen and a loss of muscle elasticity. Degenerative changes in spastic muscles could impact to the reduction of functional outcomes including muscle weakness, reduced ROM, and decreased muscle strength and contractile properties. Therefore, one obligation and goal of any rehabilitative therapy such as physical therapy, occupational therapy, and targeted exercise programs, as well as local Botulinumtoxin treatment is to prevent from or reduce chronic disability and such chronic changes in the muscle tissue [49].
In conclusion, the benefit of BoNT-A therapy generally outweight the risk of BoNT use associated degenerative changes in spastic muscles. Regular monitoring and individualized treatments can help to minimize these potential degenerative changes. Further studies in this field are warranted.
CONCLUSION
Early identification of PS-SMD has a key role in the management of PS-SMD. If physical management alone is insufficient to control the SMD with accompanying complications from increasing spastic muscle tone and involuntary phasic and tonic muscle activation, early consideration of additional treatment with BoNT-A in focal, multi-focal and segmental spasticty can help to promote effective neurorehabilitation and to avoid long-term complications. Predictors of PS-SMD, including clinical signs and brain imaging data, are established and are being ready to be applied in clinical practice. The first-line management of focal, multi-focal, and segmental PS-SMD involves comprehensive assessment, goal setting in the MPT, techniqual guided BoNT-A injections, and a setting of multimodal antispastic measures, e.g., time-adjusted adjunctive therapies like serial casting, taping, or functional electrical stimulation. Following evidence-based recommendations, multi-pattern BoNT-A therapy is safe and effective, with no severe side effects. Overall, BoNT-A treatment in the acute or early subacute phase and chronic phases of stroke can effectively reduce muscle tone and improve both passive and active functions, leading to an improved quality of life, in particular when combined with patient-centered care and multimodal or adjunctive other treatment modalities. Especially, BoNT-A injections within 3 months (early subacute phase) following stroke are strongly recommended in patients with PS-SMD or at its high risk as controlled studies with early BoNT-A interventions showed longer endurance of BoNT-A effect with lower dose per muscle and reduction in the probability to develop complications like contractures and spasticity-associated pain in the chronic phase after stroke.
 XML Download
XML Download