

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
The worldwide prevalence of obesity has nearly tripled since 1975 to 1 out of 10 adults being obese (body mass index [BMI] ≥30 kg/m2) and 4 out of 10 being overweight (BMI ≥25 kg/m2) as of 2016, according to the World Health Organization [1]. This trend of the global increase in obesity is also present in the adolescent population despite some regional disparities. On the contrary to the high-income Western countries which showed the smallest increase in adolescent obesity since 2000, the highest increase was seen in the east Asian region [2]. This trend is also prominent in the adolescents of South Korea, as well as other east Asian countries, who are exposed to sleep deprivation due to the excessive pressure to study for college entrance exams [3,4]. Such an increase in the prevalence of obesity in the adolescent population will lead to various metabolic disorders in the future which consequently carry a high risk of cardiovascular complications and mortality. Therefore, this will not only be a problem at an individual level but also a factor of serious socioeconomic burden [3].
Sleep deprivation leads to the state of chronodisruption which is known to increase obesity, risk of diabetes, cardiovascular disease, fatty liver disease, and mortality [5-11]. Previous studies have shown that sleep deprivation leads to obesity and diabetes which could also be achieved by the elimination of melatonin through the removal of the pineal gland [12]. It is also known that reversal of these negative effects could be achieved through melatonin administration in these cases [13].
Increasing evidence indicates that melatonin deficiency, due to lifestyle changes in modern society is involved in the pathogenesis of the recent worldwide increase in obesity [14,15]. Furthermore, recent discussions are highlighting the potential role of melatonin as a prevention and treatment for metabolic obesity and its complications. Hence, intensive research is being conducted to investigate the various mechanisms of its beneficial effects such as anti-inflammatory effect, insulin sensitivity, and activation of brown adipose tissues [9,16,17].
Cholesterol synthesis and cholesterol level regulation plays an important role in obesity and its complications [18-21]. Most studies have shown that melatonin inhibits cholesterol synthesis, but it is controversial whether it is 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMGCR) related [22]. In obesity development, the first step is the intestinal absorption of calories. But, only a few studies have investigated whether melatonin is involved in fat absorption [23]. In particular, there have been no studies on nutrient transporters related to lipid and glucose absorption.
Therefore, in the present study, we hypothesized that the mechanism by which melatonin suppresses obesity is due to the inhibition of cholesterol synthesis in the liver and nutrient absorption in the intestine. We evaluated the inhibitory effects of melatonin in obesity and fatty liver induced by a high-fat diet (HFD) through mRNA expression levels of proteins involved in cholesterol synthesis and reabsorption in the liver and nutrient transporters related to glucose and lipid absorption in the intestines. Additionally, we investigated whether this is a direct mechanistic effect of melatonin on hepatocytes.
Go to : 
METHODS
Animal experiments
Four-week-old male C57BL/6 mice were purchased from Orient Bio (Seongnam, Korea). The mice were housed in a humidity-controlled room maintained at 24℃ under a 12-hour lightdark cycle, with free access to different food and water. All animal experiments were performed according to the Animal Experimentation Ethics Committee of Kosin University (KMAP20-8). The mouse was divided into three groups of five mice each: (1) normal diet group (fed standard rodent chow, contains 5.54% calories from fat, 74% calories from carbohydrates, and 21% calories from protein); (2) HFD group (fed HFD that contained 60% fat, 20% protein, 20% carbohydrate); and (3) melatonin group (fed on HFD and administered melatonin). The melatonin (50 mg/kg body weight) was injected into the intraperitoneal cavity 5 days per week around the afternoon for 17 weeks. Melatonin receptors 1 (MT1) and 2 (MT2) are expressed in the liver and intestine and administration of melatonin increased the content of MT1/MT2 receptor protein in a dose-dependent manner [24,25]. Melatonin at 50 mg/kg dose had previously been shown anti-inflammatory, neuroprotective, and antiobesity effects in rats and mice [26-28].
Body weight and liver weight evaluation
Body weight was measured every week in the morning (10:00 AM to 11:00 AM). Liver weight was measured after mouse sacrifice.
Histological analysis
The liver was removed from each mouse, fixed 4% paraformaldehyde, paraffin-embedded and sectioned at 4 μm. Hematoxylin and eosin (H&E) staining was according to the standard method. Frozen sections liver, 4 µm-thick cryosections, were stained with Oil red O (Sigma-Aldrich, St. Louis, MO, USA). All sections were observed with a microscope.
Total cholesterol, apolipoprotein A1, and apolipoprotein B level analysis
All level measurement was determined in the serum. Total cholesterol level was quantified with QuickDetect total cholesterol (mouse) enzyme-linked immunosorbent assay (ELISA) kit (Bio-Vision, Milpitas, CA, USA). Apolipoprotein A1 (ApoA1) and apolipoprotein B (ApoB) levels were measured with Mouse ApoA1 ELISA kit, mouse ApoB ELISA kit (Abcam, Cambridge, UK). All kit was according to the manufacturer’s instructions.
Cell culture and treatments
The human liver cancer cell line HepG2 was cultured in 75-cm2 plastic flasks (Falcon-Becton Dickinson Labwares, Franklin Lakes, NJ, USA) and maintained at 37°C with 5% CO2 in air atmosphere in Dulbecco’s modified essential medium (DMEM) supplemented with 10% (volume/volume) heat-inactivated fetal bovine serum (FBS) and antibiotics (100 U/mL of penicillin, 100 lg/mL of streptomycin). HepG2 cells were treated to 250 or 500 μM palmitate (Sigma-Aldrich) with or without 1.5 mM melatonin conjugated to fatty acid-free bovine serum albumin (BSA) for 24 hours. HepG2 cells exposed with BSA were used as control.
Reverse transcriptase-polymerase chain reaction
Total RNA was isolated using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) and reverse transcription was performed with superscript reverse transcriptase (Invitrogen) according to the manufacturer’s instruction. Total RNA (1 μg) was used to prepare cDNA. Expression of HMGCR, low-density lipoprotein receptor (LDLR), sterol regulatory element-binding protein 2 (SREBP-2) at the liver tissue and sodium-glucose transporter 1 (SGLT1), glucose transporter 5 (GLUT5), GLUT2, Niemannpick C1-like 1 (NPC1L1) at the small intestinal epithelium were determined by polymerase chain reaction (PCR). The primers used for amplification were as follows: HMGCR forward, 5´-TGAGATCCGGAGGATCCAAG-3´ and reverse, 5´-ATCCAGCGACTATGAGCGTG-3´, LDLR forward, 5´-TTTGGAGGATGAGAACCGGC-3´ and reverse, 5´-CAGGTACTGGCAACCACCAT -3´, SREBP-2 forward, 5´-CTGGAGACCATGGAGACCCTC-3´ and reverse 5´-CAGGTACTGGCAACCACCAT-3´, SGLT1 forward, 5´-GCCATGGGTGGCTTTGAATG-3´ and reverse, 5´-GAGCAGGGACAGAACGGAAA-3´, GLUT5 forward, 5´-ACCAAGGGAGCCATCAACAAG-3´ and reverse, 5´-GTTATCCTGACTGCTGCCGTA-3´, GLUT2 forward, 5´-ACCGGGATGATTGGCATGTT-3´ and reverse 5´-GGACCTGGCCCAATCTCAAA-3´, NPC1L1 forward, 5´-CCGCGTTCCGAAACCTTGCC-3´ and reverse 5´- AGGATTGCGAAAACACCTGC-3´. Cycling parameters for the three amplifications consisted of an initial DNA denaturation at 94°C for 5 minutes followed by 35 cycles at 94°C for 20 seconds, 55°C for 20 seconds, 72°C for 20 seconds, and final extension at 72°C for 5 minutes. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as an internal control.
Quantitative polymerase chain reaction
Total RNA from mouse small intestine tissue was reverse transcribed using TOPscript cDNA Synthesis kit (Ezynomics Inc., Daejeon, Korea) following the manufactures recommendations. Quantitatve PCR was performed using the QuantStudio3 real-time PCR (Applied Biosystems, Waltham, MA, USA) and TB Green premix Ex Taq II kit according to the manufactures directions. The primers used for amplification were as follows: HMGCR forward, 5´-ATCAGCTGCACCATGCCATC-3´ and reverse, 5´-GTGCAAGCTCCTTGGAGGTC-3´, LDLR forward, 5´-AAGGACACAGCACACAACCA-3´ and reverse, 5´-CAAAGGAAGAAGACGAGGAGCAC-3´, SREBP-2 forward, 5´-CTGACTTCCCTGCTGCAGTG-3´ and reverse 5´- GTTTCCGGTGCCTCCAGAAG-3´, Expression levels for each sample, normalized to GAPDH expression, were calculated as the average of two measurements.
Immunofluorescence staining
The HepG2 cells were cultured on collagen-coated 4-well glass chamber slides in DMEM media containing 10% FBS. Cells were fixed in 4% paraformaldehyde/phosphate-buffered saline (PBS) followed by permeabilization in 0.2% Triton X-100/PBS. 3-Hydroxy-3-methylglutaryl coenzyme A (HMGCoA) was obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Appropriate primary antibodies were added to the cells for 1 hour at 37°C. For secondary labeling, cells were incubated with fluorescein-conjugated secondary antibodies (Invitrogen) for 1 hour at 37°C. Nuclei were counterstained using Deep Red (Invitrogen). Fluorescent images were observed and analyzed under fluorescence microscopy (Eclipse 80, Nikon, Tokyo, Japan).
Western blot analysis
For Western blots analysis, equivalent amounts of total protein (15 mg) were loaded onto 10% sodium dodecyl sulfate-polyacrylamide gels electrophoresis (SDS/PAGE). The gels were transferred to nitrocellulose membranes using an electro-blotting apparatus (Bio-Rad, Hercules, CA, USA) and reacted with antibodies according to standard methods. The proteins were then transferred to a nitrocellulose membrane using an electroblotting apparatus (Bio-Rad), and the membranes were incubated with each primary antibody (Anti-HMGCoA, LDLR antibodies, Santa Cruz Biotechnology). Blots were incubated with a horseradish peroxidase conjugated anti-mouse secondary antibody (Santa Cruz Biotechnology). The membranes were developed using an enhanced chemiluminescence reaction system and visualized with an Amersham Imager 600 (GE Healthcare, Chicago, IL, USA). β-Actin was used as an internal control to monitor equal protein loading.
Biochemical analysis
Plasma and liver protein levels of the pro-inflammatory cytokines interleukin 6 (IL-6), IL-10, and tumor necrosis factor α (TNF-α) were measured using commercial ELISA kits (mouse IL-6 uncoated ELISA, and mouse TNF-α uncoated ELISA, Invitrogen). All experimental steps were performed at room temperature and in duplicate. Samples were quantified by spectrophotometry at 450 nm.
Go to :
RESULTS
Melatonin attenuates HFD-induced weight gain, visceral fat accumulation, and liver steatosis
Weight gain, visceral fat accumulation, and steatosis are major contributors to metabolic obesity and its complications [29]. Therefore, we first examined these factors in mice under a normal chow diet (NCD), HFD, and HFD with melatonin administration (HFD+M) for 17 weeks. As expected, compared to the HFD mice which gained the most weight, reduced weight gain was seen in HFD+M mice while NCD mice gained the least weight (Fig. 1A). The same trend could be observed upon visual inspection of visceral and subcutaneous fat accumulation via gross examination, and extent of lipid accumulation in liver tissues via histological assessments as shown in Fig. 1B, D. In both cases, melatonin decreased fat accumulation in HFD. Liver weights, consistent with the body weight gain, were significantly heavier in HFD mice compared to the NCD mice, and melatonin administration significantly prevented liver weight gain in HFD+M mice (Fig. 1C).
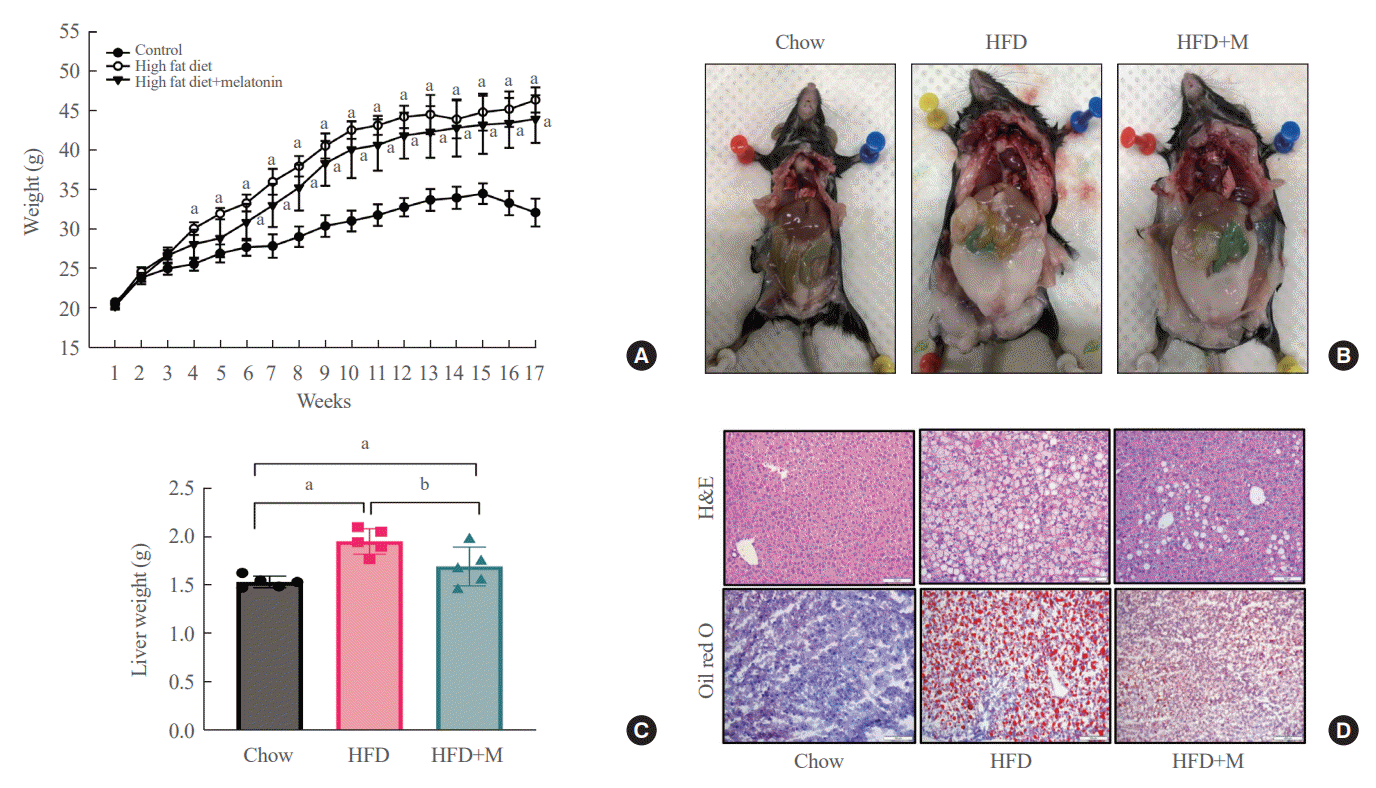 | Fig. 1.Melatonin protects against high-fat diet (HFD)-induced obesity and fat deposit in the liver. (A) body weight changes through 17 weeks, (B) gross findings of visceral fat accumulation, (C) comparison of liver weights among groups, (D) histological lipid accumulation of liver with H&E (×100) and Oil red O (×200) staining at 17 weeks. HFD+M, high-fat diet with melatonin. aP<0.05 as compared to the control; bP=0.14 as compared to the HFD.
|
Melatonin ameliorates HFD-induced elevation of lowd-ensity lipoprotein cholesterol
One of the driving forces for metabolic obesity and its complications, namely hepatic steatosis in HFD is hyperlipidemia [30]. In this aspect, serum lipid profiles were measured at 6 weeks to gain a better understanding of the mechanisms underlying the protective effects of melatonin in HFD. No significant differences were observed among the three groups in terms of total cholesterol (Fig. 2A) and ApoA1 lipoprotein levels (Fig. 2C). However, serum ApoB lipoprotein levels were significantly lower in the HFD+M mice compared to the HFD mice (Fig. 2D), and a similar trend was observed for the serum triglyceride levels as well (Fig. 2B).
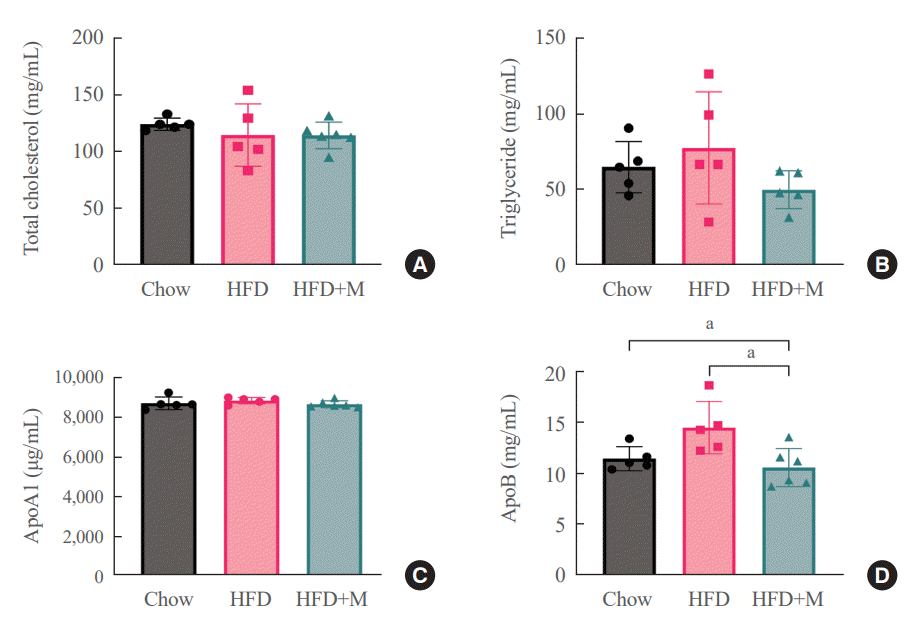 | Fig. 2.Melatonin decreased high-fat diet (HFD)-induced low-density lipoprotein cholesterol elevation. Serum lipid profile at 6 weeks (A) total cholesterol, (B) triglyceride, (C) apolipoprotein A1 (ApoA1) lipoprotein, (D) apolipoprotein B (ApoB) lipoprotein. HFD+M, high-fat diet with melatonin. aP<0.05.
|
Protective effects of melatonin from absorption of excess calories via suppression of carbohydrate and lipid transporter expressions in the intestine
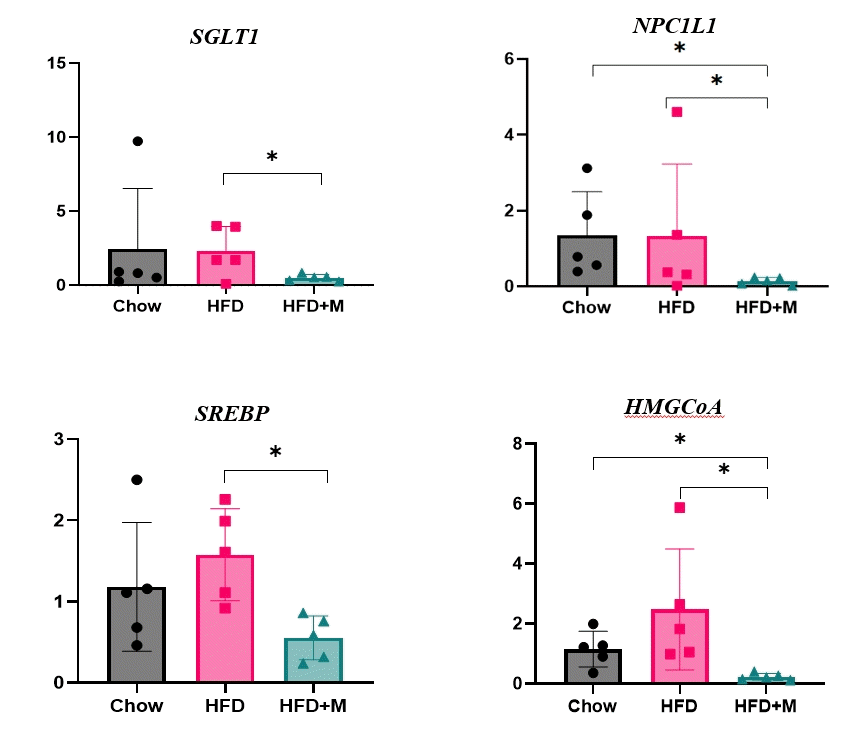
The fact that melatonin inhibits the production of chylomicrons in the intestines prompted us to investigate its effect on intestinal lipid absorption under HFD. To examine the effect of melatonin on intestinal caloric absorption, small intestinal expressions of various nutrient transporters were assessed. Interestingly, intestinal carbohydrate transporters, SGLT1 at the brush border and GLUT2 at the basolateral membranes were significantly suppressed by melatonin compared to HFD mice (Fig. 3A, C). GLUT5 at the brush border also showed a strong trend to be suppressed by melatonin compared to HFD mice (Fig. 3B). Moreover, NPC1L1, a cholesterol transporter in the liver and the intestines was also significantly suppressed by melatonin compared to HFD and NCD mice as shown in Fig. 3D.
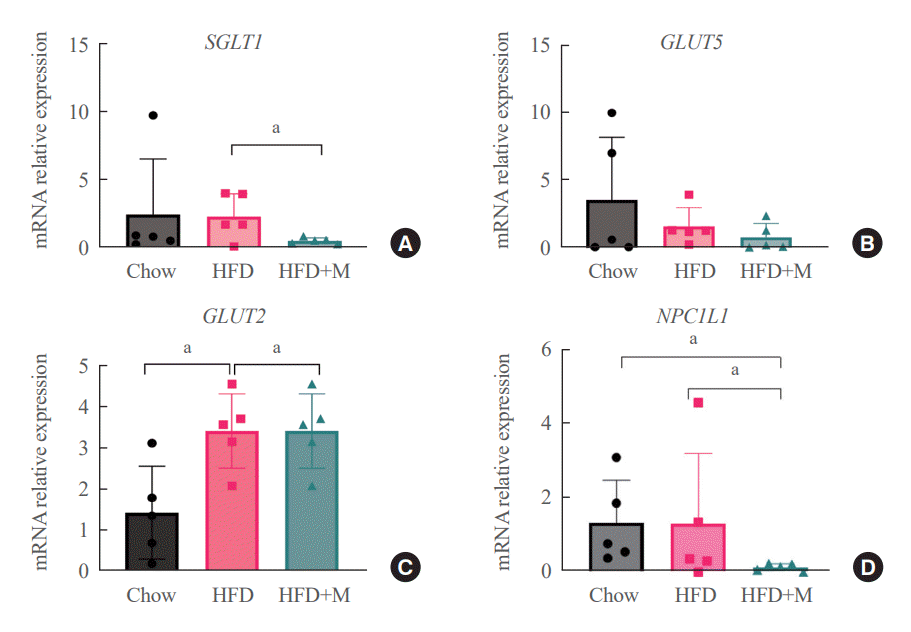 | Fig. 3.Melatonin protect excess calories absorption via suppression of carbohydrate and lipid transporters’ expression in the intestine (A) sodium-glucose transporter 1 (SGLT1) which is brush border transporter of carbohydrate, (B) glucose transporter 5 (GLUT5) which is brush border transporter of carbohydrate (C) glucose transporter 2 (GLUT2) which is basolateral membrane transporter of carbohydrate, (D) Niemann-pick C1-like 1 (NPC1L1) which is intestinal and hepatic transporter of cholesterol. HFD, high-fat diet; HFD+M, high-fat diet with melatonin. aP<0.05.
|
Melatonin suppresses hepatic cholesterol synthesis
Given the central role of the liver in the regulation of low-density lipoprotein (LDL) cholesterol levels, we further investigated the effect of melatonin on hepatic cholesterol synthesis. Therefore, we evaluated the mRNA expression levels of genes involved in hepatic cholesterol metabolism. mRNA expression levels of genes that promote cholesterol biosynthesis, SREBP-2, and HMGCR were increased by HFD [18,20]. However, melatonin administration significantly inhibited the expression levels of both genes under HFD (Fig. 4A, B). A similar pattern was seen in the expression levels of LDLR, which promotes hepatic cholesterol reuptake, although not at a statistically significant level (Fig. 4C).
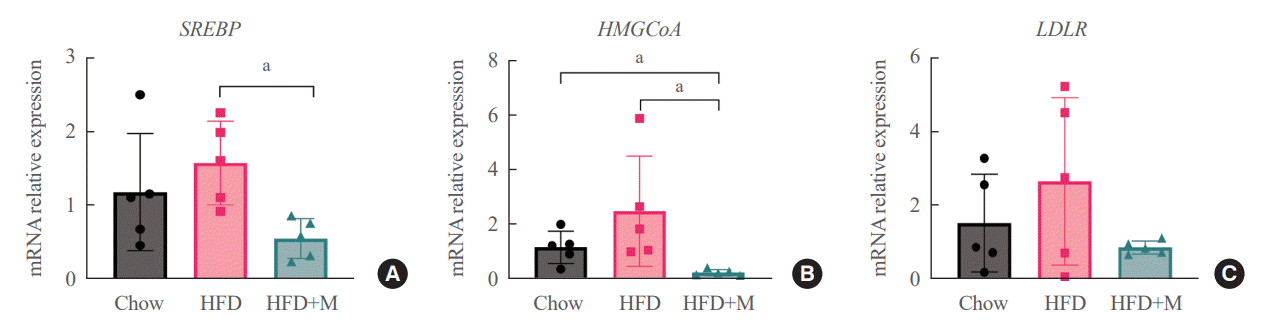 | Fig. 4.mRNA expression levels of genes involved in cholesterol metabolism in the liver. (A) sterol regulatory element-binding protein 2 (SREBP-2), (B) 3-hydroxy-3-methylglutaryl coenzyme A (HMGCoA) reductase (C) low-density lipoprotein receptor (LDLR). HFD, high-fat diet; HFD+M, high-fat diet with melatonin. aP<0.05.
|
No differences in circulatory and local inflammatory cytokine levels between groups
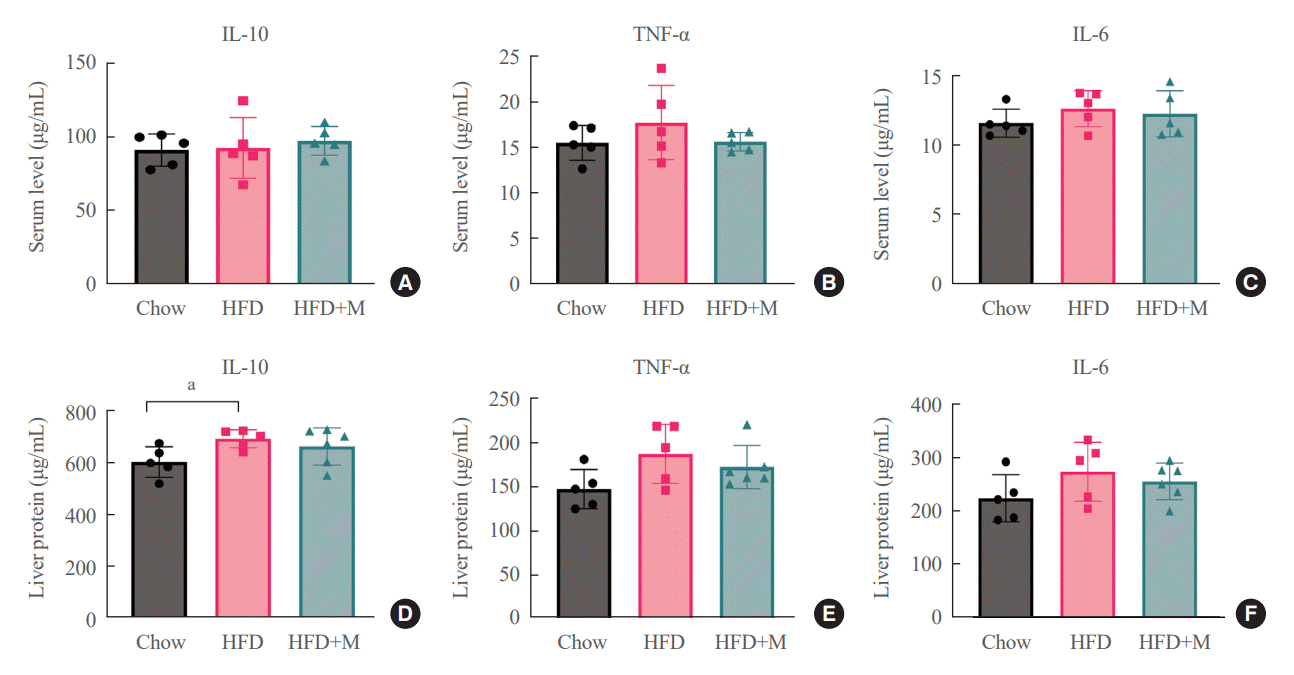
To determine whether these protective effects of melatonin on the inhibition of hepatic steatosis and obesity are through its direct action on target cells, we measured local and circulatory inflammatory cytokine levels. Measurements of local inflammatory cytokine levels were taken from the liver. As shown in Fig. 5, there were no significant differences in local and circulatory inflammatory cytokine levels between the three groups except for the liver IL-10 level, which was higher in HFD mice compared to NCD mice.
Melatonin directly inhibits palmitate-induced cholesterol synthesis in hepatocytes
Lastly, we attempted to address the potential concerns regarding the existence of other indirect pathways underlying the mechanism of action of melatonin on the hepatocytes. All hepatocytes were treated with palmitate for 6 hours, and the melatonin group was concurrently treated with melatonin for 16 hours compared to the control group which was not treated with melatonin. As a result, expression levels of SREBP-2, HMGCR, and LDLR were all decreased in the melatonin group compared to the control group as shown in Fig. 6A. We confidently demonstrated that melatonin directly inhibits cholesterol synthesis in hepatocytes through Western blot and immunofluorescence staining showing the same pattern as shown in Fig. 6B, C.
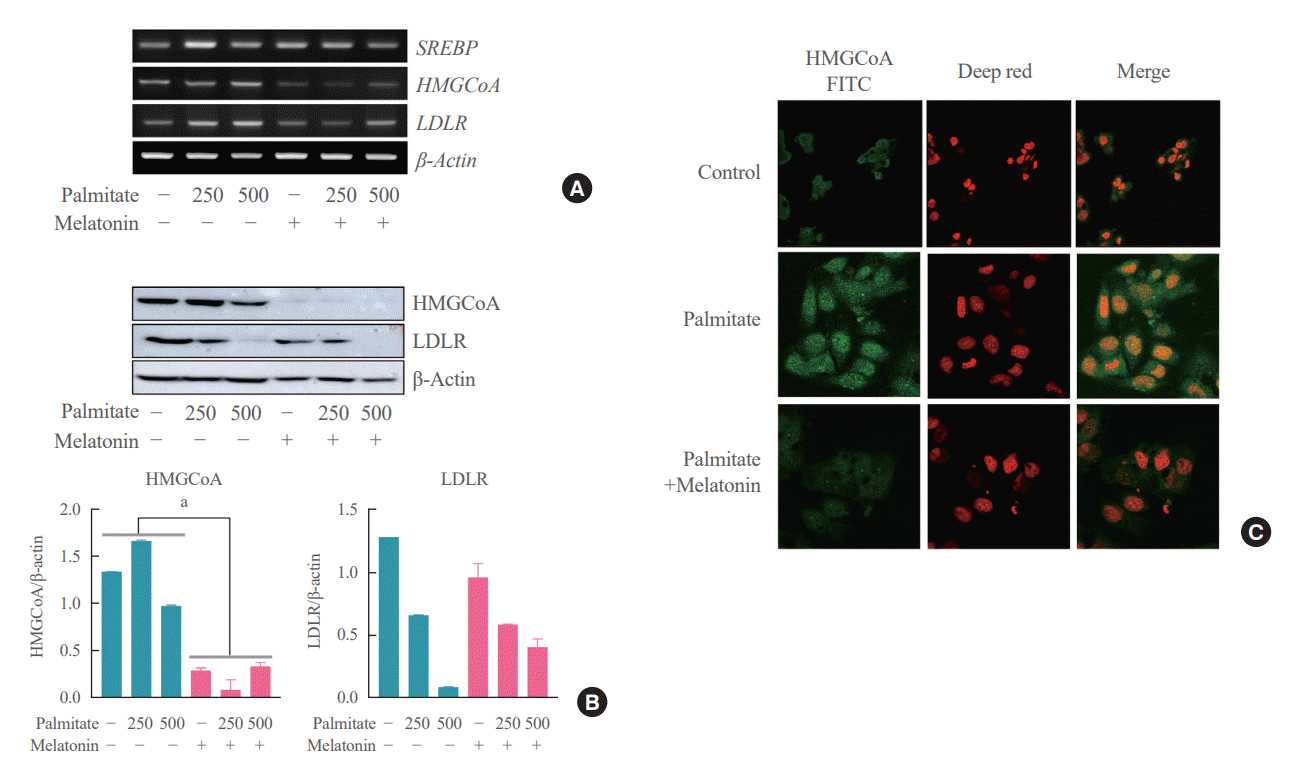 | Fig. 6.Melatonin directly inhibits palmitate-induced cholesterol synthesis in hepatocyte. (A) Hepatocytes were stimulated with palmitate for 6 hours in the absence or presence of melatonin. Levels of sterol regulatory element-binding protein (SREBP), low-density lipoprotein receptor (LDLR), 3-hydroxy-3-methylglutaryl coenzyme A (HMGCoA) were analyzed by reverse transcriptase-polymerase chain reaction. (B) Levels of LDLR, HMGCoA were analyzed by Western blot. β-Actin were used as internal loading controls. (C) Immunofluorescent images were obtained with palmitate, melatonin, and both and were compared to the control (×800). FITC, fluorescein. aP<0.01.
|
Go to :
DISCUSSION
The preventative effect of melatonin on the development of obesity and the progression of fatty liver under HFD has been well elucidated through previous studies [31,32]. The majority of these studies have identified the mechanism behind this effect in the aspect of gut microbiota modulation [17], anti-inflammatory, and anti-oxidative properties of melatonin [9,30]. However, cholesterol biosynthesis and regulation of cholesterol level is an integral part of the development of obesity and its complications [18-21] and currently, the mechanistic effect of melatonin in this aspect requires further clarity.
After 17 weeks, HFD+M mice demonstrated decreased body weight gain and visceral fat accumulation compared to HFD mice (Fig. 1A, B). Also, concurrent administration of melatonin significantly prevented weight gain of the liver compared to HFD alone (Fig. 1C). Additionally, upon histological analysis of the liver tissue using H&E and Oil red O stain, the extent of lipid accumulation was less in the HFD+M mice compared to HFD mice (Fig. 1D). Collectively, these observations indicate the preventative effect of melatonin in HFD-induced weight gain, visceral fat accumulation, and steatosis which is in alignment with previous studies.
In the current study, we sought to further explore the mechanism underlying this effect in the aspect of serum lipid profiles. In terms of total cholesterol and ApoA1 lipoprotein, no differences were seen among the three groups (Fig. 2A, C). Interestingly, the level of ApoB lipoprotein, which is a constituent of LDL cholesterol, was significantly decreased in the HFD+M mice compared to HFD mice (Fig. 2D). A similar pattern was seen for the serum triglyceride levels although not at a statistically significant level (Fig. 2B). Dietary lipid digest into cholesterol, triglyceride, and free fatty acid, and after absorption, they turn into chylomicrons to transport to the liver. The liver excretes very low density lipoprotein (VLDL) to the systemic blood, and after using triglyceride for energy fuel whole body’s LDL remains. These are ApoB-contained apolipoproteins. Whereas ApoA1 is related to high-density lipoprotein cholesterol [33,34]. Therefore, these results indicate the inhibitory effect of melatonin in production of chylomicrons in the intestines and LDL cholesterol in the liver.
Dietary cholesterol is absorbed in the intestinal epithelium, goes through esterification, and enters the circulation as chylomicrons. To date, only a few studies have reported the relationship between melatonin and fat absorption, only with regards to intracellular esterification [23]. Recent studies have highlighted the crucial role of NPC1L1 protein in the regulation of intestinal cholesterol absorption, and response toward different cholesterol concentrations [18]. Moreover, inhibition of NPC1L1 is known to prevent HFD-induced hepatic steatosis, weight gain, and induce a significant reduction in LDL cholesterol level, which is in alignment with the effect of melatonin under HFD [19]. Despite this significance of NPC1L1, the relationship between melatonin and various intestinal nutrient transporters including NPC1L1 has never been studied before. Our data for the first time highlights the inhibitory effect of melatonin on the expression of NPC1L1 protein under HFD (Fig. 3D). One considerable point is the expression of NPC1L1 was markedly downregulated in HFD+M. Because NPC1L1 is a critical mediator of cholesterol absorption, excessive suppression of NPC1L1 might have adverse effects caused from lack of cholesterol. However, the total amount of cholesterol transport via NPC1L1 was higher compared to extremely low expression of NPC 1 level as we can confirm in Fig. 2. It is speculated due to the extremely high content of fat amount (60%) of HFD compared to the chow diet which contains 5.54% of fat.
The results from our assessment of various intestinal nutrient transporters collectively indicate the fact that melatonin has an inhibitory effect on intestinal nutrient absorption under circumstances where excessive calories are supplied. Thus, highlighting the regulatory role of melatonin in intestinal caloric absorption.
The inhibitory effect of melatonin in hepatic cholesterol biosynthesis in liver has been demonstrated through previous studies [35]. However, the mechanism underlying this effect regarding the involvement of HMGCR is controversial [22]. Our examination of hepatic mRNA expression levels revealed that melatonin significantly inhibited the HFD-induced elevation of SREBP-2 and HMGCR, which promotes cholesterol biosynthesis (Fig. 4A, B). In a similar pattern, decreased expression of LDLR was noticed although not at a statistically significant level (Fig. 4C). We assume that this decrease in expression of LDLR is secondary to the decreased amount of cholesterol biosynthesis.
Melatonin is known to exert some of its effects mediated through modulation of inflammatory cytokines or cellular interleukin responses rather than direct action on target cells [36]. Our data showed no significant differences in local and inflammatory cytokine levels between HFD and HFD+M mice (Fig. 5), which suggests the aforementioned effects of melatonin are the result of its direct action on target cells rather than indirectly mediated through circulatory factors. Additionally, we performed an in vitro cell experiment through a previously established cellular hepatic model of human hepatocytes in steatosis by treating HepG2 cells with palmitate [37,38]. Our data demonstrated decreased expression levels of SREBP-2, HMGCR, and LDLR (Fig. 6) which is consistent with our in vivo findings in mouse hepatocytes. Despite the controversial result in another in vitro study [22], our collective evidence from in vitro and in vivo mouse experiments indicates the direct action of melatonin on hepatocytes inhibiting cholesterol biosynthesis.
This study has several limitations. First, since there are only five numbers in each group, several results were not reached statistical significance. In terms of inflammatory cytokines, the lack of significance in the HFD group might be due to the insufficient number. Second, we didn’t perform protein levels. The expression of mRNA could represent the protein expression, sometimes it may not coincide with protein expression. Although most papers related to intestinal nutrient transporters showed mRNA expression only, future studies are needed to determine how melatonin affects protein levels and activities.
The significance of our study is uncovering the anti-obesogenic and steatoprotective mechanism of melatonin under HFD with regard to cholesterol absorption and biosynthesis. Especially, our study is the first to discover the inhibitory effect of melatonin in intestinal lipid absorption through suppression of NPC1L1, which is known to have anti-obesogenic and steatoprotective properties [19]. Moreover, we did not expose our mice to sleep deprivation to maintain the normal diurnal profile of melatonin. Our results were obtained under this circumstance which implies the therapeutic potential of melatonin as a treatment for obesity and fatty liver. Indeed, it must be noted that blood levels of melatonin were not measured in our study. Additional studies with measurements of higher serum melatonin levels in HFD+M mice and its diurnal fluctuations would further strengthen these results.
In conclusion, our data demonstrate the inhibitory effect of melatonin in weight gain and progression of fatty liver in mice exposed to 17 weeks of HFD through suppression of lipid absorption via decreased intestinal NPC1L1 expression and decreased cholesterol synthesis.
Go to :
 XML Download
XML Download