

 PDF
PDF Citation
Citation Print
Print




INTRODUCTION
Sudden cardiac arrest is defined as the sudden cessation of cardiac activity with no sign of normal respiration and circulation, which is a medical emergency that progresses to sudden cardiac death (SCD) if not managed immediately.1) Ischemic heart disease is the most common and serious burden predisposing patients to SCD, followed by cardiomyopathy.2)3) Inherited arrhythmia (IA) is a primary electric disorder caused by genetic alterations in cardiac ion channel proteins and comprises various inherited diseases such as Brugada syndrome (BrS), long QT syndrome (LQTS), and idiopathic ventricular fibrillation (IVF). IA is rare cardiac cause of SCD, and is more prevalent in the Asian population.4)5)6)
There has been difficulty in the clinical diagnosis of IA in patients with insufficient electrical evidence. Advances in genetic testing methods have led to the identification of unrevealed genotypes, and have shown potential as diagnostic and prognostic tools for inherited cardiovascular disease. In other words, genetic testing using a multi-gene panel might provide a molecular diagnosis of underdiagnosed SCD, which is promising in the diagnosis of IA.
Although the significance of IA is more accentuated in Asian populations, little is known about its clinical characteristics and genetic background in Asian populations. To date, most genetic studies in Asian IA have been based on Japanese cohorts, and a multicenter registry with a large volume of IA probands is lacking. Therefore, based on a multicenter Korean IA cohort, we aimed to investigate the clinical characteristics of Korean IA probands and further analyze the genetic findings via next-generation sequencing (NGS).
METHODS
Ethical statement
This study was approved by the institutional review board of each hospital (IRB No. 2014AN0104), and informed consent was obtained from all patients.
Study population
This study was based on a multicenter cohort of the Korean IA Registry, which included 401 probands and family members in 14 tertiary centers from February 2014 to November 2017 (Supplementary Table 1).
The inclusion criteria of IA probands conformed to the Heart Rhythm Society/European Heart Rhythm Association/Asia Pacific Heart Rhythm Society expert consensus statement on the diagnosis during study period, and the inclusion was confirmed by an electronic case report form to standardize patient inclusion from different centers.7) In our cohort, IA included seven diseases: BrS, LQTS, short QT syndrome (SQTS), catecholaminergic polymorphic ventricular tachycardia (CPVT), early repolarization syndrome, IVF, and arrhythmogenic right ventricular cardiomyopathy (ARVC). Probands were excluded if they had 1) significant coronary artery disease, 2) systemic disease such as malignancy with a life expectancy of less than one year, and 3) cardiomyopathy except ARVC. Family members of probands (siblings, parents, or children) were enrolled if they had not been diagnosed with IA previously and agreed for enrollment and data collection for the study. Eight participants were excluded because of missing data, and the registry comprised 265 IA probands and 128 family members.
Data collections
Clinical data were collected using an electronic case report form. The data consisted of baseline characteristics, including age at the time of diagnosis, sex, family history of IA and SCD, body mass index, symptoms or signs at diagnosis, history of ventricular tachycardia or ventricular fibrillation, and underlying comorbidities. Further clinical data including electrocardiographic and echocardiographic findings, provocation test results (epinephrine and sodium channel blocker), current medications, and implantable cardioverter-defibrillator (ICD) implantations were also collected. Genetic testing was recommended for all patients. Those without informed consent for genetic analysis and its expenses were excluded for further genetic testing.
Genetic testing using next-generation sequencing panel
Among the 265 probands, genetic testing by NGS was performed on 216 probands. Genomic DNA was extracted from peripheral blood leukocytes using the Wizard Genomic DNA Purification Kit (Promega, Madison, WI, USA). A Celemics Customized Target Enrichment Kit (Celemics, Seoul, Korea) was used for library preparation. Sequencing was performed using the hybrid capture method by generating 2×150 base pairs paired-end reads on the Illumina MiSeq platform (Illumina Inc., San Diego, CA, USA). Sequencing was performed using 174 multigene panels designed by Illumina Design Studio (Illumina Inc.) to investigate the 3,130 target lesions known to cause cardiovascular disease according to the Human Gene Mutation Database (HGMD, http://www.hgmd.cf.ac.uk) (Supplementary Table 2).
Alignment of sequence reads, indexing of the reference genome, hg19 (GRCh37), and variant calling were conducted with a pipeline based on GATK Best Practice (alignment was performed with the BWA-mem [version 0.7.12], duplicated reads were marked with Picard [version 1.96, http://picard.sourceforge.net] and local alignment, base quality recalibration, variant calling was performed with the Genome Analysis Tool kit [GATK, version 3.2-2], and annotation was performed with VEP [Variant Effect Predictor], dbNSFP v3.02.).
The data were analyzed in the following three steps. The primary analysis was sequence generation, which converts sequenced raw data into a text-based format (FASTQ) to store both biological sequences and the corresponding quality scores. Secondary analysis involved sequence processing with variant calling, which selects a different sequence (variant) compared with the reference sequence. Tertiary analysis included validation and clinical interpretation by database and algorithms (evidence-based, frequent-based, functional, and predictive).8)
Bioinformatics
Filtering of false positive variants was performed as follows: the region coding the actual protein from the flanking region was identified; only loss of function variants (frameshift, nonsense, consensus splice site) were selected in order to distinguish mutations causing the actual variant; variants with allele frequency of more than 0.3 were included; minor allele frequency of more than 5% in population frequency database was excluded; compound heterozygous variants in autosomal recessive inheritance genes were included; and the disease-causing mutation reported only in HGMD was included.
The following criteria were used for validation of analysis accuracy: read depth was at least 10X or higher for increased sensitivity, variants with 10X coverage of 90% or more were included, the number of calling variants was at least 100 (per batch).
Multiple databases were used to analyze the variants. The variant allele frequency and minor allele frequency were determined according to the following population databases:1000 Genome Project (http://www.internationalgenome.org/), Exome Aggregation Consortium (ExAC, http://exac.broadinstitute.org), single nucleotide polymorphism database (dbSNP, https://www.ncbi.nlm.nih.gov/SNP/), and Korean reference genome database (KRGDB, http://coda.nih.go.kr/coda/KRGDB/index.jsp). The association with IA was determined using disease databases, including HGMD, Clinvar (https://www.ncbi.nlm.nih.gov/clinvar/), and OMIM (http://www.omim.org/). Different in-silico analysis tools including SIFT (https://sift.bii.a-star.edu.sg/), Polyphen2 (HDIV/HVAR) (http://genetics.bwh.harvard.edu/pph2/), likelihood-ratio test, Mutation taster/assessor (http://www.mutationtaster.org/, http://mutationassessor.org/), FATHMN (http://fathmm.biocompute.org.uk), PROVEAN (http://provean.jcvi.org) were harnessed to determine the probabilities of variant pathogenicity.
Variant classifications
Variants were classified into five categories (pathogenic, likely pathogenic, variant of unknown significance, likely benign, or benign) based on the Standard and Guidelines for the Interpretation of Sequence Variant of American College of Medical Genetics and Genomics and the Association for Molecular Pathology.9) Genotypes were defined as positive if the following three analytic criteria were met: 1) variant is in disease-associated gene according to American College of Medical Genetics and Genomics classification,10)11) 2) variant allele frequency is more than 0.3, and 3) minor allele frequency is less than 5%. Genetic yield was defined as ratio of probands detected with positive genotypes, and was further specified as ‘yield of pathogenic or likely pathogenic (P/LP) variants.’ Variant analysis and interpretation were curated by clinical cardiologists and genetic bioinformatics specialists.
Statistical analysis
Categorical variables were described as numbers and percentages, and continuous variables were presented as means and standard deviations. For comparison of categorical and continuous variables, Student’s t-test, Mann-Whitney U test, χ2 test, and Fisher’s exact test were used as indicated. One-way analysis of variance (ANOVA) was used to compare the three variables. All tests were two-tailed, and a p-value of ≤0.05 was considered statistically significant. All statistical analyses were performed using SPSS software (version 26; IBM Corp., Armonk, NY, USA).
RESULTS
The Korean IA Registry included 401 probands diagnosed with IA or family members of those with IA. Eight cases were excluded because of lack of clinical information. Among the 393 cases, 265 were probands with IA and 128 were family members (Figure 1).

Clinical characteristics
The distribution of IA in the probands is shown in Figure 2. IVF and BrS was the most prevalent diseases, diagnosed in 96 cases (36.2%) and 95 cases (35.8%), respectively. Other diseases included LQTS (n=54, 20.4%), ARVC (n=9, 3.4%), early repolarization syndrome (n=7, 2.6%), CPVT (n=3, 1.1%), and SQTS (n=1, 0.4%).
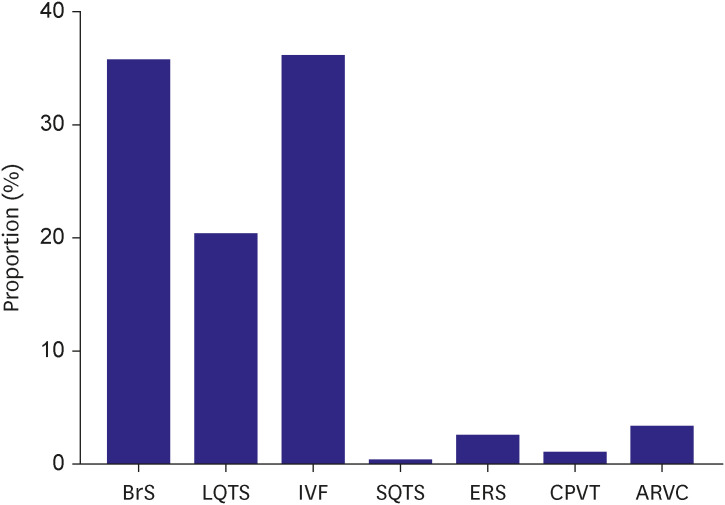
Figure 2
Proportion of inherited arrhythmia in Korean probands.
ARVC = arrhythmogenic right ventricular cardiomyopathy; BrS = Brugada syndrome; CPVT = catecholaminergic polymorphic ventricular tachycardia; ERS = early repolarization syndrome; IVF = idiopathic ventricular fibrillation; LQTS = long QT syndrome; SQTS = short QT syndrome.
![]()
The baseline characteristics of IA probands are presented in Table 1. Mean age of onset was 40.4±14.2 years, and 73.6% were male. Most of the probands had disease-related symptoms, with previous aborted SCD history being the most common presentation (80.8%). Family history of SCD was not frequent (16.6%), and echocardiography showed preserved left ventricular ejection fraction (58.4±10.1%). A total of 209 patients (78.9%) underwent ICD implantation. The incidence of comorbidities is shown in Supplementary Figure 1.

Table 1
Baseline characteristics of inherited arrhythmia probands
Values are presented as mean ± standard deviation or number (%).
The baseline characteristics of inherited arrhythmia probands and comparisons of Brugada syndrome, long QT syndrome, and idiopathic ventricular fibrillation are described.
BrS = Brugada syndrome; ICD = implantable cardioverter-defibrillator; IVF = ventricular fibrillation; LQTS = Long QT syndrome; SCD = sudden cardiac death; VF = ventricular fibrillation; VT = ventricular tachycardia.
![]()
The three most frequent disease entities, BrS, LQTS, and IVF, were compared based on their clinical characteristics (Table 1). The mean age of onset and left ventricular ejection fraction did not differ among the diseases. Significant difference in sex distribution was seen in three disease entities: while BrS was mostly composed of male probands (92.6%) as well as IVF (82.3%), LQTS showed higher proportion of female probands (77.8%). Clinical presentation was also different; BrS and LQTS showed a lower incidence of ventricular fibrillation and aborted SCD than IVF (p<0.001). Family history of SCD was more frequent in BrS, with relatively low incidence in IVF (26.3% vs. 7.3%, p=0.001). Majority of patients with IVF received ICD implantation (88.5%), whereas those with LQTS had undergone relatively higher proportion of medical therapy (87.0%).
Genetic testing
Genetic testing using the NGS panel was performed on 216 probands (Figure 1). Sixty-nine probands (31.9%) were genotype-positive with 88 variants, which was further narrowed down to 14 probands (6.4%) with 14 P/LP variants (Table 2). Clinical characteristics were compared between the genotype positive and genotype negative probands (Table 3). No significant difference was noted in demographic and clinical characteristics except in those with left ventricular ejection fraction, which was significantly lower in genotype positive probands (n=69) compared to genotype negative probands (n=147) (54.7±11.3 vs. 59.3±9.2%, p=0.005).


Table 2
Pathogenic or likely pathogenic variants of inherited arrhythmia probands
Fourteen probands were detected with 14 pathogenic or likely pathogenic variants. SCA indicates cardiac arrest not specified as shockable rhythm, such as ventricular fibrillation or pulseless ventricular tachycardia.
ACMG = American College of Medical Genetics, ARVC = arrhythmogenic right ventricular cardiomyopathy; BrS = Brugada syndrome; HCM = hypertrophic cardiomyopathy; IVF = idiopathic ventricular fibrillation; LQTS = long QT syndrome; SCA = sudden cardiac arrest, VF = ventricular fibrillation.
![]()
Table 3
Comparison of clinical characteristics between genotype positive and negative probands
Values are presented as number (%) or mean ± standard deviation.
Clinical characteristics of 216 consecutive IA probands with genetic data are compared.
ICD = implantable cardioverter-defibrillator; SCD = sudden cardiac death; VF = ventricular fibrillation; VT = ventricular tachycardia.
![]()
Brugada syndrome
Among 95 patients with BrS, 82 underwent genetic testing and among these, 16 (19.5%) were genotype positive (Supplementary Tables 3 and 4). The most commonly detected genotype was SCN5A (n=9; 10.9%). However, the minor allele frequency of SCN5A was relatively high in the Korean population database (KRGDB); p.Arg1193Gln was classified as a common variant rather than a disease-associated variant. Other genotypes were also detected: two CACNA1C, two SCN1B, two HCN4, one CACNB2, one PKP2, and one SCN3B. Two rare variants of SCN5A, p.Val789Ile and p.Val281Met, were defined as likely pathogenic and pathogenic variant, respectively.
Long QT syndrome
Genetic testing was performed in 42 of 54 LQTS probands. Ten cases (23.8%) were detected with genotypes (Supplementary Tables 5 and 6). After excluding three patients with the common variants of SCN5A (p.Arg1193Gln) mentioned previously in BrS, KCNQ1 and KCNH2 were the most common variants. The other genotypes included SCN5A, ANK2, and AKAP9. Two variants were P/LP variants (one KCNQ1 and one KCNH2), both of which were novel and not found in any population database. These were predicted to have deleterious effects on silico analysis.
Idiopathic ventricular fibrillation
Genetic testing was performed on 74 IVF probands, and among these, 40 probands (54.0%) had 57 genotypes (Supplementary Tables 7 and 8). Notably, IVF probands had diverse genetic backgrounds that included not only IA but also cardiomyopathy. IA-associated genetic variants were detected in 15 patients accounting for 17 genotypes (SCN5A, SCN1B, KCNE1, ANK2, KCNH2 and RYR2). Thirty-one patients had cardiomyopathy-associated genetic variants, accounting for 40 genotypes (DSP, PKP2, DSG2, TMEM43, DSC2, JUP, TTN, MYPN, TMPO, SGCD, MYBPC3, MYH7, MYH6, TNNI3, NEXN). Several patients had multiple variants and six patients had both channelopathy- and cardiomyopathy-related genotypes. The most common genotype relevant disease was ARVC (19 out of 57 genotypes), followed by hypertrophic cardiomyopathy (14 out of 57 genotypes). Among the IA-related variants, BrS was the most common related disease (six out of 57 genotypes). Notably, most of the P/LP variants were associated with cardiomyopathies, which implies a significant probability of concealed cardiomyopathy in IVF probands. Namely, nine out of 10 patients with P/LP variants had cardiomyopathy-related variants. Six IVF patients were detected with hypertrophic cardiomyopathy-related variants that encode sarcomere proteins (MYBPC3, MYH7, TNNI3) and three patients had ARVC-related variants (DSG2, DSP, JUP), mostly located in desmosome proteins. None of the IVF probands had IVF-related genotypes, such as DPP6 or CALM1.
Other inherited arrhythmias
Few genotypes were detected in other IAs. One proband with SQTS, six with early repolarization syndrome, and three with CPVT underwent genetic testing, but only single nucleotide polymorphism was detected, without the detection of significant genotypes. Eight of the nine ARVC probands received genetic testing. Three patients were genotype positive (PKP2, DSG2, and TTN) but had no P/LP variants (Supplementary Tables 9 and 10).
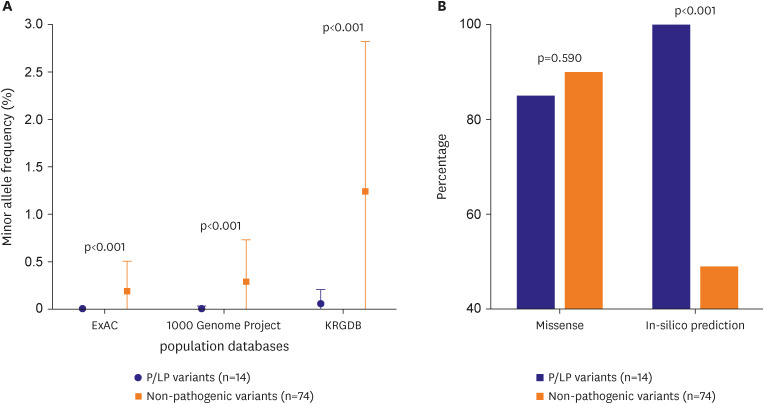
Comparison of pathogenic or likely pathogenic vs. non-pathogenic
The genetic characteristics of P/LP and non-pathogenic variants were further compared among the 88 detected genetic variants (Figure 3). There was no significant difference in proportion of missense mutations between non-pathogenic (n=74) and P/LP variants (n=14) (90.5%, 85.7%, respectively, p=0.590), and deleterious effect predicted by in-silico analysis was less prevalent in non-pathogenic variants (49.2%, 100.0%, respectively, p<0.001). Although the degree varied among three population databases, P/LP variants were less frequent in general population compared to non-pathogenic variants (Minor allele frequency, ExAC: 0.192 vs. 0.004%, p<0.001; 1000 Genome Project: 0.288 vs. 0.007%, p<0.001; KRGDB: 1.244 vs. 0.058%, p<0.001).
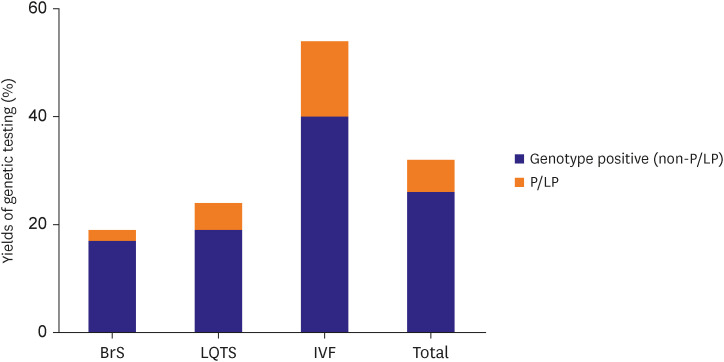
Yield of genetic testing with next-generation-sequencing panel
The yield of genetic testing using the NGS panel is shown in Figure 4. Positive genotypes and P/LP variants were found in in 31.9% and 6.4% of total probands, respectively. IVF probands had the highest genetic yield (54.0% for positive genotype and 13.5% for P/LP variant), followed by LQTS (23.8% for positive genotype and 4.8% for P/LP variant). Among the three disease entities, BrS showed lowest genetic yield (19.5% for positive genotype and 2.4% for P/LP variant), which was significantly lower compared with IVF (p=0.014).
DISCUSSION
Based on the multicenter IA registry of Korea, this study investigated clinical characteristics and further assessed the genetic background of IA probands using an NGS panel. Major findings of this study can be summarized as follows: First, the most common disease entity was IVF, followed by BrS, and LQTS. Second, certain differences in clinical characteristics were demonstrated among the three disease entities, including sex differences and lethal arrhythmic events. Third, there was significant difference of left ventricular ejection fraction between positive and negative genotype probands (54.7±11.3 vs. 59.3±9.2%, p=0.005). Lastly, the genetic testing yield was relatively low for BrS and LQTS; however, IVF revealed a higher genetic testing yield, which was supported by a substantial proportion of cardiomyopathy-related variants.
IVF, BrS, and LQTS are the three most common disease entities, which accounted for more than 90% of whole Korean IA population. The disease was usually diagnosed in the fourth decade, with a slightly earlier onset of LQTS. In addition, BrS and IVF were predominant in males and LQTS in females. Compared to BrS or LQTS, the IVF subgroup encountered significantly more lethal arrhythmic events, which led to a higher incidence of ICD implantation. This may be explained by the current diagnostic criteria for each disease, where electrocardiographic abnormality is the major diagnostic criterion for BrS and LQTS, whereas an event of unexplained cardiac arrest is required to diagnose IVF.
Clinical characteristics of probands were compared depending on the presence of genotypes; there was no difference between genotype positive and genotype negative probands except for those with left ventricular ejection fraction. The exact mechanism for the lower left ventricular ejection fraction in the positive genotype remains unclear, but it might be partly explained by concept of concealed cardiomyopathy in IVF, since IVF probands resulted highest genetic yield.12)13) In other words, clinically overt cardiomyopathy might be preceded by a preclinical phase, and sudden cardiac arrest might be the first cardiac manifestation in this population. Nevertheless, this finding should be interpreted with caution. The numerical differences of left ventricular ejection fraction between two groups were observed by simple comparison, and do not address direct cause-and-effect relationship with genetic result. In addition, these differences were not substantial (54.7±11.3 vs. 59.3±9.2%). Although lower left ventricular ejection fraction in positive genotypes may not alter treatment strategy in clinical practice, it may support consistent monitoring of structural change as well as possibility of progression to clinical cardiomyopathy in genotype positive probands. Advanced research specified in each disease entity is needed to fully interpret this finding.
In our study, genetic yield for via the NGS panel was found to be lower than that reported in previous studies.14)15) Although SCN5A was the most common genotype detected in BrS probands, the genetic yield of SCN5A was 10.9% (9 out of 82 probands), which was lower than that reported previously reported (approximately 20%). Non-SCN5A variants (CACNA1C, SCN1B, HCN4, CACNB2, PKP2, and SCN3B) were also prevalent and mostly rare. However, only two SCN5A variants were identified as P/LP variants, which is consistent with previous evidence that SCN5A is the only definitive molecular marker for BrS.16)
In LQTS, more than 75% probands are reported to have three major genes (KCNQ1, KCNH2, SCN5A) in western population and in a Japanese cohort.17)18) In previous studies of Korean LQTS probands, LQTS genes were detected in 50–70% of probands.19)20) The NGS panel utilized in this study included all LQTS-related genes that have definite or strong evidence for causality of LQTS except CALM3.
21) In addition, our study utilized a larger genetic panel, but genetic testing including multiple genes did not improve the diagnostic yield in the LQTS subgroup. There may be several reasons for relatively low yield in LQTS. First, the NGS panel used in our study was developed based on a western genetic background, which may not be fully reflect genetic background of Asian IA probands. Second, thorough segregation study of familial genetic screening was not done in every proband, which have weakened the pathogenicity classified by American College of Medical Genetics and Genomics. Third, there may be selection bias that led to overdiagnosis of LQTS. LQTS is likely to be overdiagnosed due to clinical symptom such as vasovagal syncope, and transient QT interval prolongation during stress condition.22) In our data, LQTS revealed higher incidence of syncope as well as lower incidence of aborted SCD, which implies possibility of overdiagnosis in LQTS.
In contrast to BrS and LQTS, the genetic yield of IVF was substantial in our study. More than half of the patients were detected with certain genotype, and 13.5% had P/LP variants. Our findings were consistent with previous studies in Caucasian IVF population that revealed 10–18% of cardiomyopathy-related P/LP variants.23)24) A high incidence of IVF indicates a paucity of clinical phenotypes to identify the cause of ventricular fibrillation or SCD at the initial examination. In addition, the higher genetic detection rate in IVF is mainly due to the cardiomyopathy-related P/LP variant found in this specific group. ARVC exhibits a preclinical phase that does not express evident structural abnormality.25) Similarly, hypertrophic cardiomyopathy may not be diagnosed at initial phase, if left ventricular wall thickness is equivocal or in borderline range.26) Our finding implies the possibility of concealed cardiomyopathy in this population, which suggests serial imaging follow-up, such as echocardiography, in IVF probands to undercover disease progression to overt cardiomyopathy.
Therefore, regarding the genetic yield of NGS panels in Korean IA probands, genetic testing using a large-volume gene panel did not provide better yield in BrS and LQTS. On the other hand, in IVF, multiple genetic sequencing, such as NGS or whole-exome sequencing, may enable molecular diagnosis of concealed phenotypes, which can change future management strategies, such as further imaging studies or additional familial screening.
This study was based on the first multicenter registry of Korean IA probands and utilizes a large genetic testing panel including 174 genetic variants of cardiovascular disease. It provided comprehensive information on the clinical and genetic characteristics of the three most common disease entities of IA. More studies are needed in Korean IA probands in order to improve our understanding of the genetic mechanism, and further establish genotype-phenotype correlation in IVF to identify concealed cardiomyopathy.
This study had several limitations. First, the epidemiologic distribution of this registry may not represent the actual prevalence of each disease in Korea. Although IA probands were enrolled regardless of symptoms, the registry resulted high proportion of sudden cardiac arrest survivor (80.8% in total probands), which might lead to selection bias that increase proportion of IVF. It may have resulted from the clinical need of genetic testing in sudden cardiac arrest of unknown origin (or IVF) to identify genetic susceptibility to specific disease. Second, it focused on more common diseases of IA and provides limited information on other rare diseases, such as early repolarization syndrome, CPVT, and SQTS. Considering its prevalence, further studies with larger numbers of sudden cardiac arrest survivors are needed to encompass the rare diseases of IA. Third, clinical information may be limited due to the retrospective nature of the data. Most of the clinical information was obtained at the time of sudden cardiac arrest or admission, which may reflect worse cardiac function (left ventricular ejection fraction) or cardiac conduction property. In this regard, further collection of follow-up data might supplement this shortcoming. Lastly, although many cardiomyopathy-related genotypes were found in IVF, IVF-related rare variants such as DPP6 or CALM1 were not identified in this population. However, this study provides important insights into genetic backgrounds of IA in Korea although further study for genetic data including rare IAs.
BrS IVF, and LQTS were three major disease entities in Korean IA probands, which differed not only in their clinical characteristics but also in the arrhythmic event rate that led to ICD implantation. Genotype positive probands showed lower left ventricular ejection fraction compared to genotype negative probands. The genetic yield of BrS and LQTS was lower than previous studies, even with the use of the large-volume NGS panel. In IVF probands, genetic screening for cardiovascular disease-associated genes may be promising to unveil hidden phenotype such as concealed cardiomyopathy.
 XML Download
XML Download