

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
The interest in adipose tissue has increased in recent years because of the rise in global rates of obesity and associated comorbidities [1]. While the earlier studies of adipose tissue focused primarily on its energy-storing function, it has since been recognized that adipose tissue is a complex and dynamic organ composed of many cell types, including adipocytes, immune cells, endothelial cells, blood cells, and others, altogether contributing to the maintenance of metabolic health [2]. Adipose tissue modulates systemic metabolism by taking up glucose and fatty acids and by secreting a variety of bioactive molecules such as hormones, metabolites, and genetic materials. Dysfunctional adipose tissue can promote the development of cardiometabolic disorders and conversely, targeted manipulation of adipose tissue function has the potential to confer metabolic benefits.
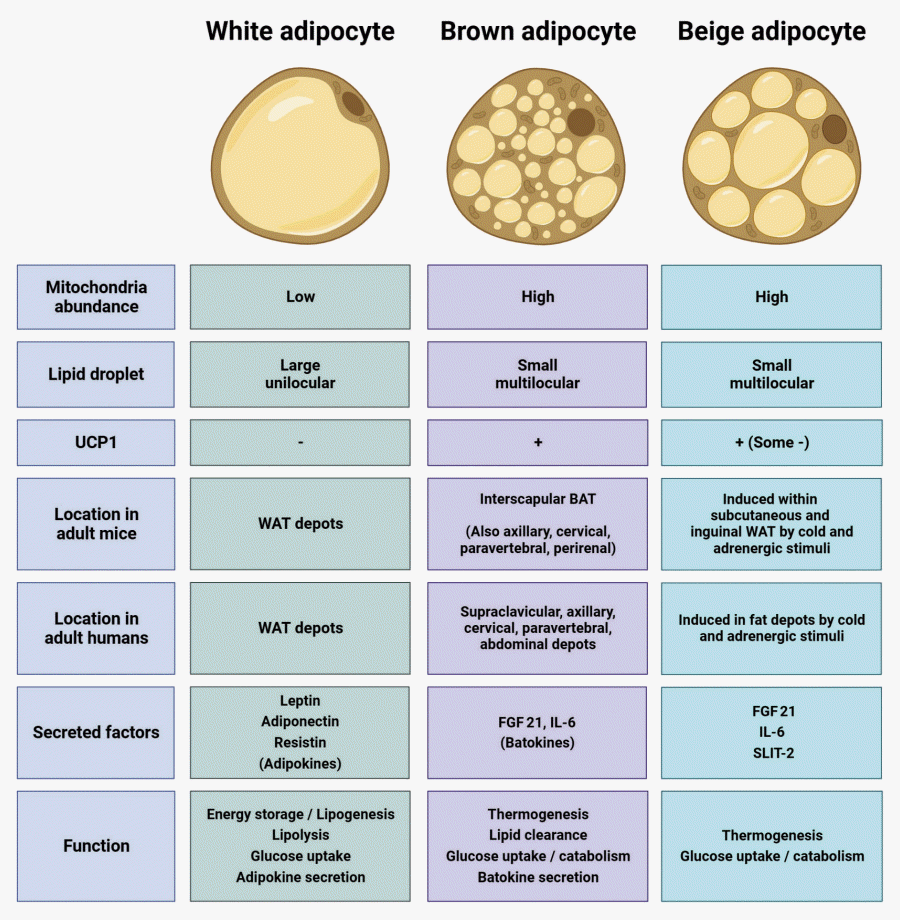
White adipose tissue (WAT) is the most common type of adipose tissue and is the site where majority of the excess calories are stored. Brown adipose tissue (BAT) has a characteristic brown colored appearance that distinguishes it visually from WAT (Fig. 1). This brown color is due to a greater abundance of iron-rich mitochondria in BAT and reflects the ability of BAT to produce heat in response to environmental stimuli [3]. Consistent with this thermoregulatory function, BAT is especially abundant in animals that are subjected to cold stress, such as those that are postnatal, living under severe weather conditions, or undergoing hibernation. In C57BL/6J mice, BAT is most prominent in the interscapular area. Although BAT accounts for less than 1% of the body weight in adult mice [4], compared to about 15% for WAT, adult mice are still dependent on BAT for survival during acute cold exposure. In humans, BAT has been reported to make up about 5% of the body weight in neonates [5] and is identified in the interscapular region at birth. While human BAT was previously thought to be detectable only in infants and Arctic indigenous peoples, functional imaging studies have demonstrated the presence of BAT depots in adult humans at multiple sites including the supraclavicular, thoracic and abdominal regions [6-9]. A key difference between BAT and WAT is the expression of uncoupling protein 1 (UCP1), an inner mitochondrial membrane protein that uncouples the mitochondrial proton gradient from adenosine triphosphate (ATP) production in BAT to generate heat. Brown-like adipocytes capable of carrying out thermogenesis, termed beige adipocytes, have been described and are thought to arise either by recruitment of a specific preadipocyte subpopulation within WAT or by direct conversion of white adipocytes to beige adipocytes in response to appropriate environmental stimuli [3]. Beige adipocytes have a molecular signature that is distinct from classical brown adipocytes. They express UCP1 but have also been reported to utilize UCP1-independent thermogenic mechanisms such as futile creatine cycling and Ca2+ cycling [10-13]. UCP1-negative beige adipocytes that possess thermogenic capacity based on creatine cycling have been described [14]. Development of thermogenic adipocytes within WAT, sometimes called “beiging” or “browning,” has been observed in humans with conditions accompanied by heightened adrenergic stress such as chronic cold exposure or burn trauma [3,15,16]. Supraclavicular BAT in adult humans appears to contain both brown and beige adipocytes, as molecular markers associated with both cell types are found [17]. It is likely that there exists still a far greater degree of adipocyte heterogeneity than is currently understood [18]. Ample evidence already indicates that both thermogenic and non-thermogenic adipocytes are key players in the regulation of metabolic health, as further discussed below.
ADIPOSE TISSUE AND CARDIOMETABOLIC DISEASES
Physiology
Adipose tissue is the primary location for the storage of energy in mammals. Under conditions of energy surplus, adipocytes synthesize triglycerides from free fatty acids released from circulating triglyceride-rich lipoproteins such as chylomicrons and very-low-density lipoproteins via the action of lipoprotein lipase (LPL) in the capillary [19]. Sequential esterification of fatty acids to glycerol, which comes from glucose, results in the formation of triglycerides. The terminal and committed step of triglyceride synthesis is catalyzed by diacylglycerol acyltransferase (DGAT). In addition to taking up fatty acids from the blood, adipocytes can also utilize fatty acids converted from acetyl coenzyme A (acetyl-CoA) within the cell, a process known as de novo lipogenesis. Under normal conditions, de novo lipogenesis in WAT is thought to be minor compared to that in liver but can assume a greater importance on a high-carbohydrate diet. Acetyl-CoA carboxylase 1 (ACC1) and fatty acid synthase are key rate limiting enzymes in de novo lipogenesis.
When energy intake chronically exceeds energy expenditure, adipose tissue expands by increasing the size of individual adipocytes (hypertrophy) and through the formation of additional adipocytes recruited from the preadipocyte population (hyperplasia). This results in obesity and can be associated with extensive tissue remodeling, activation of inflammation, and systemic metabolic dysfunction such as insulin resistance [2,20]. The location of adipose tissue is highly relevant; accumulation of visceral fat produces adverse health consequences, whereas subcutaneous fat is considered metabolically healthy or neutral [21]. In lipodystrophy, subcutaneous adipose tissue is decreased or absent, causing deposition of lipid in visceral adipose tissue and at ectopic sites such as liver, insulin resistance, dyslipidemia, and a higher risk of cardiometabolic disease [22].
At times of energy deprivation or increased demand, triglycerides stored in adipocytes are broken down into fatty acids by the sequential catalytic actions of adipocyte triglyceride lipase (ATGL), hormone-sensitive lipase (HSL), and monoacylglycerol lipase. Catecholamines stimulate lipolysis through cyclic adenosine monophosphate (cAMP) signaling and protein kinase A (PKA) activation, which in turn targets ATGL and HSL, whereas insulin inhibits lipolysis by activating phosphodiesterase 3B and converting cAMP into 5’ adenosine monophosphate [23]. Defective insulin signaling in adipocytes causes high rates of basal lipolysis and increased circulating levels of free fatty acids and glycerol, which leads to lipid accumulation at ectopic sites such as liver and muscle and interference with insulin signaling at those sites [24]. Thus, the maintenance of normal levels of lipogenesis and lipolysis by adipocytes is critically important in systemic metabolism.
Adipose tissue also plays an essential role in glucose homeostasis. Although skeletal muscle is the tissue mainly responsible for insulin-sensitive glucose uptake (80% to 85%), adipose tissue also expresses the insulin-regulated glucose transporter type 4 (GLUT4) and participates in peripheral glucose disposal [25]. In addition, thermogenic adipose tissue can serve as a glucose sink under conditions that produce adrenergic stimulation [26]. Mice that selectively lack GLUT4 in adipose tissue have normal adipose tissue mass but develop glucose intolerance and hyperinsulinemia [27]. Surprisingly, adipocyte-selective deletion of the insulin receptor improves glucose tolerance despite the impairment of glucose uptake in adipose tissue [28]. This has been attributed to chronic compensatory changes taking place in other insulin-responsive tissues of the conventional GLUT4 knockout mice. An inducible knockout of GLUT4 in adipose tissue, which does not allow sufficient time for such compensatory responses, produces insulin resistance [29]. This suggests that adipose tissue controls glucose metabolism in other metabolic tissues and implicates secreted factors, genetic materials, and metabolites such as free fatty acids.
Adipose tissue has been described to release over 50 known hormones and proteomics studies suggest many more [30,31]. Three of the most extensive studied adipokines are leptin, adiponectin, and resistin. Leptin levels correlate well with adipose tissue mass and are regulated by food intake [32]. Leptin has anorexic effects and increases energy expenditure [33]. Obese states are usually characterized by leptin resistance and hyperleptinemia, however, limiting the therapeutic utility of exogenous leptin in the context of obesity. A recent study suggested that lowering leptin levels in obesity may be beneficial [34]. Adiponectin enhances muscle glucose uptake and suppresses hepatic glucose production, and also has anti-inflammatory effects [35]. Resistin, which derives its name from the fact that it mediates insulin resistance, has been associated with a proinflammatory effect and a higher risk of cardiometabolic diseases [36]. Administration of recombinant resistin to normal mice impaired glucose tolerance and neutralization of resistin with an antibody reversed diet-induced insulin resistance [37]. While the function of resistin is conserved across species, human resistin is thought to be secreted primarily by adipose tissue macrophages while mouse resistin is secreted by adipocytes themselves [38]. Adipose tissue also produces numerous other substances that possess endocrine, paracrine, or autocrine effects, including adipsin, retinol binding protein 4 (RBP4), bone morphogenetic proteins, fibroblast growth factor 21 (FGF21), vascular endothelial growth factor A (VEGF-A), plasminogen activator inhibitor 1 (PAI-1), apelin, omentin, aprosin, vaspin, and visfatin [30]. There has been much recent interest in exosomes, or membrane-bound vesicles released into the extracellular space, which can contain lipids, proteins, RNA, and metabolites derived from adipocytes or targeting adipocytes [39]. Exosomal microRNAs (miRNAs) originating from adipose tissue have been shown to regulate gene expression in distance tissues such as liver and essentially function as adipokines [40]. In one study, adipose exosomes from high-fat diet-fed mice caused hippocampal and cortical synaptic damage and cognitive impairment, and miR-9-3p in the exosomes was implicated [41]. This was considered a potential explanation for cognitive impairment associated with metabolic syndrome.
Mechanisms
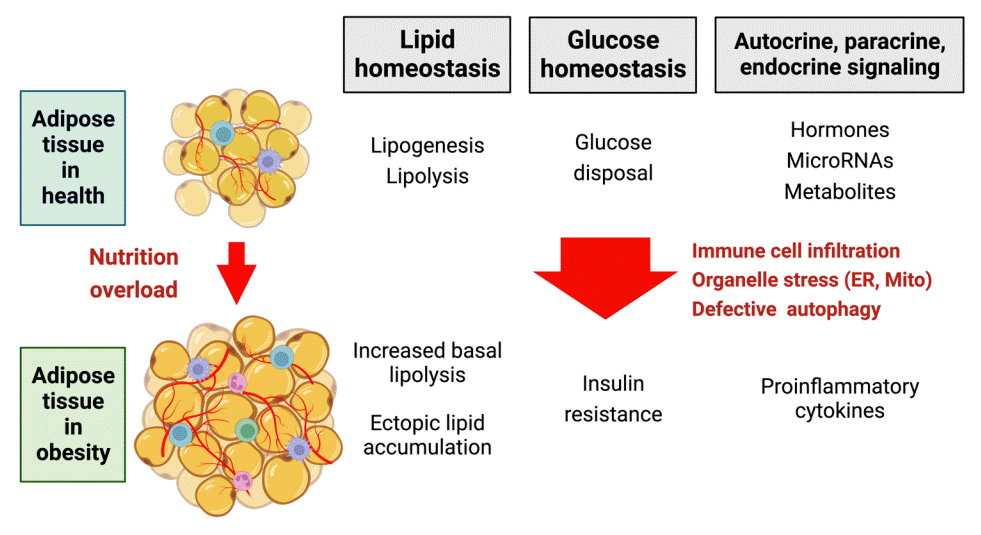
Adipose tissue dysfunction in obesity can predispose to metabolic disease by multiple mechanisms including inflammation, increased circulating free fatty acid levels and ectopic lipid accumulation, and alteration of the adipose tissue secretome (Fig. 2). Adipose tissue undergoes extensive remodeling in obesity, which encompasses not just an increase in the size and number of adipocytes but also tissue infiltration by immune cells, tissue hypoxia, accumulation of extracellular matrix components, and organelle-level dysfunction in adipocyte endoplasmic reticulum (ER), mitochondria, and lipid droplets. It has been suggested that, when the physiological capacity of WAT to accommodate excess lipid is exceeded, this triggers organelle stress and associated maladaptive responses [42]. Some studies, however, indicate that adipocyte turnover or adipocyte size do not correlate with metabolic health in a simple manner [43].
Inflammation has been studied extensively in the context of the pathogenesis of insulin resistance. Mice placed on a highfat diet induce inflammatory genes in adipose tissue, associated with immune cell infiltration [44]. Macrophages are estimated to account for under 10% of adipose tissue cells in lean mice but over 50% in genetically obese ob/ob mice [45]. The recruited macrophages are proinflammatory in character and conventionally classified as M1 type macrophages, whereas the M2 type macrophages resident in normal adipose tissue are anti-inflammatory and protective [46]. The macrophage phenotype switch from M2 to M1 in adipose tissue is associated with increased secretion of proinflammatory mediators and serves to propagate chronic tissue inflammation. Recent studies using single-cell RNA-sequencing have revealed evidence of greater heterogeneity in macrophage subpopulations than the simple M1/M2 switch model [47]. In addition, novel macrophage subtypes such as metabolically activated macrophages and oxidized macrophages have been reported to be involved in insulin resistance [46]. Besides macrophages, T-cells in adipose tissue also undergo phenotypic changes in obesity [48]. Recently, a specific population of B-cells known as T-bet-expressing (T-bet+) B-cells has been shown to increase in obesity and secrete the proinflammatory chemokine C-X-C motif chemokine ligand 10 (CXCL10), with detrimental metabolic consequences [49]. In obesity, adipocytes and immune cells residing in adipose tissue secrete many proinflammatory factors including tumor necrosis factor α (TNFα) and interleukin 6 (IL-6), which impact adipose tissue function and produce systemic effects via activation of stress kinases to promote insulin resistance [50,51]. For example, TNFα activates c-Jun N-terminal kinase, which phosphorylates insulin receptor substrate-1 (IRS-1) and blocks insulin signaling in muscle cells [52]. TNFα also affects multiple aspects of adipose tissue biology such as lipogenesis and lipolysis, adipogenesis, thermogenesis, mitochondrial function, and ER stress [53].
Inflammasomes, which are multiprotein complexes expressed in immune cells and serve as intracellular sensors for innate immunity, have emerged as significant mediators of adipose tissue inflammation associated with metabolic dysfunction [54]. The Nod-like receptor protein 3 (NLRP3) inflammasome is activated by various metabolic stress signals including proinflammatory lipids like ceramides, excessive Ca2+ efflux from the ER, and mitochondrial damage and oxidative stress. Downstream effectors of the NLRP3 inflammasome such as interleukin 1β (IL-1β) impair adipocyte insulin signaling and act to lower glucose uptake and lipogenesis, while increasing lipolysis [55,56]. A high-fat diet is associated with increased activation of the inflammasome and IL-1β in adipose tissue, while genetic deletion of NLRP3 in mice protects from insulin resistance and hepatic steatosis associated with diet-induced obesity [56].
The delicate balance between lipogenesis and lipolysis is disrupted in obesity due to adipose tissue inflammation and elevated levels of cytokines such as TNFα that interfere with insulin signaling. Obesity and insulin resistance are characterized by high basal rates of lipolysis, which in turn increase circulating free fatty acid levels [57]. Certain fatty acids have been shown to activate stress signaling pathways, produce mitochondrial dysfunction and oxidative stress, or lead to ectopic lipid accumulation in metabolic tissues such as muscle, liver, and pancreatic islets [24]. Deposition of ectopic fat in insulin-responsive metabolic tissues generally has an inhibitory effect on insulin signaling, a phenomenon commonly referred to as lipotoxicity.
Abnormalities in cellular metabolism associated with obesity include ER stress, mitochondrial dysfunction, and defects in mitophagy and autophagy, which can stimulate stress signaling pathways that trigger inflammation and recruit immune cells to adipose tissue [42]. A high-fat diet causes ER stress, or a state of abnormal protein folding, possibly in part because of changes in membrane lipid composition and dynamics [58]. Conversely, ER stress may contribute to the development of obesity, forming a vicious cycle [59,60]. ER stress activates the unfolded protein response (UPR), which has three ER transmembrane proteins acting as key sensors, namely protein kinase R-like endoplasmic reticulum kinase (PERK, also known as EIF2AK3), inositol-requiring enzyme 1α (IRE1α, also called ERN1), and activating transcription factor 6 [61]. The UPR serves to restore ER homeostasis (adaptive UPR) or initiate programmed cell death in case of severe and sustained ER stress (maladaptive UPR). While adaptive UPR is considered protective, maladaptive UPR is proposed to have a pathogenetic role in obesity. Ablation of IRE1α in myeloid cells from mice lessened diet-induced obesity and insulin resistance [59].
Nutrient overload can lead to mitochondrial dysfunction possibly because the tricarboxylic acid cycle and the electron transport chain (ETC) become saturated, reducing the ability to regenerate oxidized nicotinamide adenine dinucleotide (NAD+), and also increasing the production of reactive oxygen species [42,62]. Studies have described reduced levels of mitochondrial amount or function in adipose tissues of obese human subjects [63-65]. Whether mitochondrial dysfunction in adipocytes causes metabolic disease has not been clearly determined. Adipose-specific deletion of mitochondrial transcription factor A (TFAM) under the adiponectin promoter led to widespread adipocyte death [66]. To investigate the phenotype of a milder mitochondrial defect, adipose-specific knockout of NADH:ubiquinone oxidoreductase subunit S4 (Ndufs4), one of the subunits in mitochondrial ETC complex I, was created and demonstrated an increased susceptibility to develop diet-induced weight gain, glucose intolerance, and adipose tissue inflammation [67]. Interestingly, young male mice but not young females exhibited this propensity, suggesting a sexual dimorphism in this regard. In many mouse models of diet-induced metabolic dysfunction, female mice are relatively protected, and it has been suggested that this may be due to intrinsic differences in male and female mitochondria [68]. A recent systems genetics study provided support for this notion by demonstrating that in both humans and mice, adipose tissue mitochondrial content and function associate with body mass index (BMI) and insulin resistance in a sex-dependent manner [69]. No such association was found with mitochondria from other tissues, indicating a unique role for adipose tissue mitochondria in the regulation of systemic metabolism.
Human studies
Extensive literature exists that explores the link between adipose tissue and human metabolic health [70]. In humans, gluteofemoral fat is a prominent example of a subcutaneous WAT depot, whereas omental, mesenteric, and retroperitoneal fat are examples of visceral WAT [71]. This differs from mice, in which inguinal fat is often the largest subcutaneous WAT depot and perigonadal (epididymal or periovarian) fat and retroperitoneal fat are the largest visceral WAT depots. As noted above, subcutaneous WAT is viewed as metabolically healthy, while increased visceral WAT mass has been identified as a risk factor for cardiovascular disease, type 2 diabetes mellitus (T2DM), and steatohepatitis [72,73]. Visceral fat correlates with markers of inflammation and insulin resistance [74]. To explain the differences between the metabolic effects of subcutaneous WAT and visceral WAT, it has been suggested that visceral WAT releases metabolites into the portal vein to impact hepatic metabolism, unlike subcutaneous WAT that bypasses the portal vein [75]. However, surgical removal of omental fat did not confer metabolic benefits in randomized controlled trials, whether performed by itself or in conjunction with bariatric surgery [76,77]. Removal of subcutaneous WAT by liposuction also had neutral effects on metabolism [78].
Obese individuals are often said to have metabolically healthy obesity (MHO) if they have no more than two out of the five components of metabolic syndrome, which consist of increased waist circumference, high triglyceride level, low high-density lipoprotein cholesterol level, hypertension, and high fasting glucose [70]. Others have recently proposed a more detailed set of criteria encompassing the absence of cardiometabolic disease, healthy cardiometabolic profile, normal blood pressure, normal intrahepatic triglyceride content, and normal insulin sensitivity [79]. The reported prevalence of MHO varies from 6% to 60% of adults with obesity. However, individuals with MHO often still have some features of metabolic syndrome and about half eventually progress to metabolically unhealthy obesity (MUO) when followed for 10 years [80]. While individuals with MHO and MUO have similar percentage of total body fat, those with MHO have less visceral WAT and less intrahepatic triglyceride deposition than MUO [81]. Adipose tissue from people with MHO are different not only in the relative distribution of fat depots, but also in having a greater capacity for lipogenesis in subcutaneous abdominal fat, less adipose tissue fibrosis, less adipose tissue macrophages in both subcutaneous and visceral abdominal fat, and higher adiponectin levels [79,82,83]. No differences between MHO and MUO were seen in adipocyte proliferation rates and conflicting results were reported regarding mean adipocyte size, basal lipolysis rates, and plasma free fatty acid levels.
The mainstay of obesity therapy is behavioral modification consisting of dietary changes and moderate daily exercise [84]. Bariatric surgery is invasive but effective and produces longterm metabolic benefits including reduced risk of cardiovascular mortality. Most available pharmaceutical interventions for obesity are appetite suppressants that reduce food intake [85]. Orlistat, which is an inhibitor of gastric and pancreatic lipases causing dietary fat malabsorption, is the only exception. Among the anorexiants, glucagon-like peptide-1 (GLP-1) receptor agonists not only target the brain to lower the appetite but also stimulate insulin secretion from pancreatic β-cells to improve glucose metabolism [86]. Adipose tissue GLP-1 receptor expression has been detected [87] and thus GLP-1 receptor agonists may have direct actions on adipose tissue, besides working via the central nervous system. GLP-1 receptor agonists have been associated with favorable metabolic changes in obese human subjects such as lowering of BMI, improved ability of insulin to suppress lipolysis and hepatic glucose production, and reduction in hepatic steatosis, and have been shown to reduce the risk of cardiovascular morbidity and mortality in people with T2DM [88-90]. Prolonged treatment with GLP-1 receptor agonists have also been reported to activate thermogenic fat in humans [91]. A dual GLP-1 and gastric inhibitory peptide (GIP) receptor co-agonist was recently approved by the U.S. Food and Drug Administration (FDA) for the treatment of T2DM and has also been shown to be effective for the treatment of obesity, with most patients achieving more than 20% weight loss in a 72-week trial [92]. GIP has been reported to enhance lipid clearance by enhancing triglyceride storage in WAT, increase WAT blood flow, stimulate glucose uptake, and lower free fatty acids in humans [93,94]. The GIP receptor is expressed in human adipocytes and in both subcutaneous and visceral WAT [95]. Because of the addition of GIP, the dual receptor agonist targets adipose tissue more effectively and holds promise for the treatment of obesity and associated metabolic dysfunction.
THERMOGENIC ADIPOSE TISSUE AND SYSTEMIC METABOLISM
Physiology
In thermogenic fat, β3-adrenergic signaling is considered the dominant signaling pathway controlling thermogenic activity [96]. Sympathetic nervous stimulation triggered by environmental cues causes the release of norepinephrine, which binds to β3-adrenergic receptors present on the surface of white and brown adipocytes. Elevated levels of cAMP that result from β3-receptor stimulation promote the activation of PKA, which in turn phosphorylates downstream targets such as p38 mitogen activated protein kinase (MAPK), cAMP response element binding protein (CREB), and HSL. Phosphorylation of p38 and CREB activates the thermogenic gene program [97]. Phosphorylation of HSL promotes lipolysis, releasing fatty acids that bind and directly activate UCP1 in thermogenic adipocytes [98]. However, lipolysis within BAT itself is not required for thermogenesis [99]. Current data suggest that BAT primarily utilizes blood glucose and circulating free fatty acids released from WAT lipolysis as substrates for thermogenesis [99,100]. BAT is also involved in the clearance of plasma triglycerides in a manner dependent on LPL, which hydrolyzes triglycerides into fatty acids and glycerol, and transmembrane receptor CD36, which facilitates the uptake of fatty acids into BAT [101]. BAT activity may thus have a major lipid-lowering effect. In mice, BAT is estimated to account for nearly half of ingested triglycerides taken up by tissues after a meal [101]. Activation of BAT by the β3-adrenergic agonist CL326,243 reduced hypercholesterolemia and protected mice from atherosclerosis [102].
In addition to systemic lipid homeostasis, BAT has an important role in glucose metabolism. Stimulation of adrenergic signaling by cold exposure induces the transcription of the glucose transporters GLUT1 and GLUT4 via the canonical cAMP pathway and increases their translocation to the plasma membrane of brown adipocytes via mammalian target of rapamycin pathway [103]. Other genes involved in glucose uptake and catabolism are also upregulated. Glucose uptake by BAT is also regulated by insulin signaling, which acts via the phosphoinositide 3-kinase-phosphoinositide-dependent kinase-1-protein kinase B (PI3K-PDK1-Akt) pathway and promotes translocation of GLUT4 to the plasma membrane. In cold-exposed obese mice, BAT was found to be responsible for nearly 75% of the glucose uptake [101]. For these reasons, BAT has been termed a “glucose sink” or a “metabolic sink” [26]. The therapeutic potential of BAT has been highlighted by transplantation experiments in which subcutaneous transplants of embryonic BAT leads to euglycemia in the streptozotocin mouse model of type 1 diabetes mellitus [104]. In another study, BAT transplants from donor mice into the visceral cavity of age- and sex-matched recipient mice resulted in improved glucose tolerance, increased insulin sensitivity, and a reversal of high-fat diet-induced insulin resistance [105]. Transplantation of human pluripotent stem cell-derived brown adipocytes into mice has also been reported to improve systemic glucose tolerance and lipid metabolism [106].
Similar to WAT, BAT is known to secrete an array of endocrine, paracrine, and autocrine factors which exert regulatory effects [107,108]. The molecules secreted by BAT, often referred to as batokines, overlap only partially with the adipokines produced by WAT. Some well-known adipokines such as adiponectin are also important batokines, but others such as leptin are secreted at a much lower level in BAT compared to WAT [109]. FGF21 is one of the earliest batokines to be characterized and has received attention because of its pleiotropic effects on multiple metabolic tissues and its promise as a possible therapeutic agent against metabolic diseases [110]. FGF21 causes weight loss, enhances systemic glucose disposal, and improves lipid profile. While FGF21 is produced mainly by the liver, it is also secreted by BAT upon thermogenic activation [111]. FGF21 may be involved in BAT-to-liver signaling since it can target the liver itself and potentially offer protection from the development of fatty liver disease [112]. Administration of FGF21 to liver-specific insulin receptor knockout mice increased glucose uptake in BAT, promoted browning of WAT, and increased overall energy expenditure [113]. Mice deficient in FGF21 had an impaired ability to convert WAT to beige fat [114]. Thus, liver-derived FGF21 can participate in thermogenic adipose tissue development and function.
Another batokine that has been studied is IL-6, which is secreted from BAT in response to stress and is required for adaptive “fight or flight” responses and hyperglycemia via induction of hepatic gluconeogenesis [115]. Stress-induced secretion of IL-6 is mediated through β-adrenergic signaling but does not require UCP1 or activation of the thermogenic program. BAT transplants from IL-6 deleted mice failed to replicate the metabolic benefits of wild-type BAT transplants, indicating that IL-6 is necessary for the systemic effects of BAT activation [105]. IL-6 is thus a candidate for mediating BAT-to-liver communication. Given that IL-6 is classically known as a cytokine, it may also mediate the effects of BAT on inflammation [116]. Other batokines that target immune cells include CXCL14, growth/differentiation factor 15 (GDF-15), and lipid-derived molecules such as 12-hydroxyeicosapentanoic acid and 12,13-dihyroxy-9Z-octadecenoic acid (12,13-diHOME) [107]. 12,13-diHOME is released from BAT following cold exposure and exercise training and has been shown to enhance BAT thermogenesis in a paracrine fashion and increase cardiac function and cardiomyocyte respiration [117-119]. BAT also produces myostatin (GDF-8), which acts on skeletal muscle to modulate the exercise capacity, and neuregulin 4, which has been implicated in hepatic lipogenesis and atherosclerotic progression [120,121]. Other BAT-derived molecules including growth factors, polypeptides, lipid metabolites, and genetic materials can serve as batokines. Circulating miRNAs released from BAT such as exosomal miRNA-99b has been suggested to target the liver to control hepatic FGF21 [40]. Therefore, BAT has essential physiological roles in the regulation of adaptive thermogenesis, lipid and glucose homeostasis, as well as crosstalk with other metabolic organs to modulate systemic metabolism.
Mechanisms
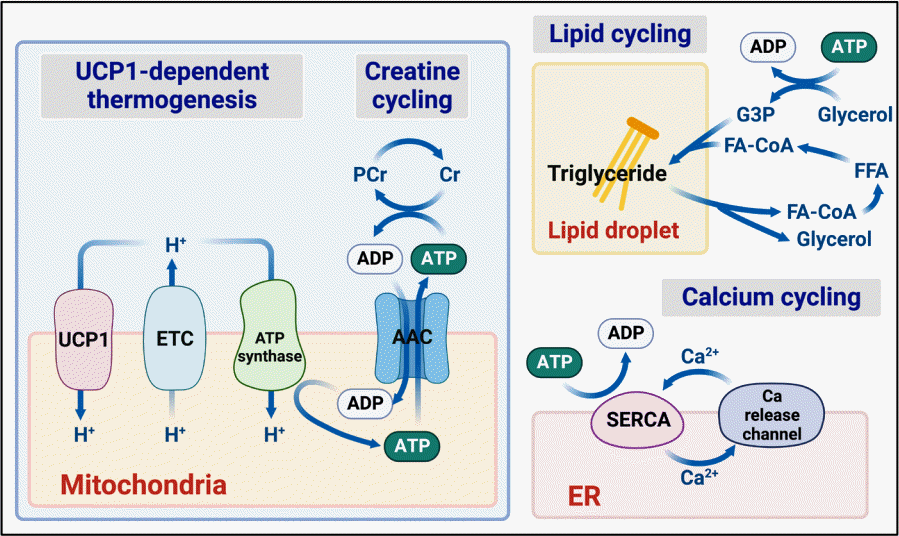
In principle, a better understanding of the regulatory pathways that control fat thermogenesis may lead to new approaches to enhance thermogenic activity. As mentioned above, BAT relies primarily on UCP1, which dissipates the proton gradient across the inner mitochondrial membrane and uncouples electron transport from ATP synthesis (Fig. 3). UCP1 activation thereby increases the flow of oxidative substrates through the ETC and drives thermogenic respiration. Besides UCP1-dependent thermogenic mechanisms, beige adipocytes can also utilize UCP1-independent mechanisms that rely on ATP-consuming futile cycles of substrates [10-13]. Such cycles involve biochemical reactions running in opposite directions so that there is no net change in the substrate concentrations but ATP is still consumed, resulting in heat generation. Mice that lack UCP1 die from hypothermia when acutely exposed to cold temperatures but survive if ambient temperature is lowered incrementally, indicating the existence of UCP1-independent thermogenic mechanisms [122]. Such mechanisms may be operative in BAT, beige adipocytes that are recruited during chronic cold adaptation, or in other tissues. Paradoxically, despite the absence of thermogenesis via mitochondrial uncoupling, UCP1 null mice are not more susceptible to weight gain than wild-type mice on a high-fat diet, even when housed at thermoneutrality (30°C) [123]. In contrast, mice that are deficient in all three β-adrenergic receptors are predisposed to diet-induced obesity [124]. Based on these observations, it has been suggested that UCP1-independent pathways triggered by β-adrenergic signaling likely underlie the obesity-resistant phenotype of UCP1 null mice [123]. UCP1 is activated by long-chain free fatty acids and inhibited by purine nucleotides [98]. The expression of the UCP1 gene is controlled by transcriptional regulators including PR domain containing 16 (Prdm16), EBF transcription factor 2 (Ebf2), peroxisome proliferator-activated receptor γ coactivator-1 α (PGC1α), zinc finger protein 423 (Zfp423), and zinc finger protein 516 (Zfp516), and epigenetic regulators such as mixed leukemia lineage 3/4 (Mll3/4), lysine-specific demethylase 1 (Lsd1), and Jumonji C domain-containing histone demethylase 2a (Jhdm2a) [125,126]. Adipose deletion of Prdm16 in mice leads to a failure to develop subcutaneous beige fat following cold exposure or β3-adrenergic agonist treatment [127]. Ebf2 deficiency causes BAT developmental abnormalities although this is eventually compensated for in adult mice [128]. Recently, loss of the mitochondrial protein Letm1 domain containing 1 (Letmd1) was shown to produce severe abnormalities in BAT mitochondria and the expression of thermogenesis related genes, including UCP1, and caused the knockout mice to die from hypothermia upon cold exposure, resembling the UCP1 null mice [129,130]. β3-Adrenoreceptor-stimulated increases in energy expenditure are completely abolished in these mice. Consistent with the idea that Letmd1 modulates UCP1 abundance, the physiological expression of Letmd1 correlates highly with the UCP1 expression and BAT thermogenic activity.
Creatine-dependent substrate cycling is a mitochondrial futile cycle that increases thermogenic respiration by pairing the forward and reverse phosphotransfer reactions of creatine and phosphocreatine (PCr). Upon β3-adrenergic signaling, mitochondrial creatine kinase (Mi-CK) in the mitochondrial intermembrane space phosphorylates creatine to produce PCr and hydrolyzes ATP. In beige fat under adenosine diphosphate (ADP)-limited conditions, phosphorylation of creatine stimulates a substrate cycle of mitochondrial ATP turnover and enhances ADP-dependent thermogenic respiration [10,14]. This mechanism has also been reported to be operative in human and mouse brown adipocytes [10,131]. Several genes related to creatine metabolism such as the Mi-CK isozymes mitochondrial creatine kinase 1 (CKMT1) and CKMT2 and the global creatine transporter solute carrier family 6 member 8 (SLC6A8) were found to be increased in UCP1 null mice, presumably as a compensatory response [10]. Genetic inactivation of glycine amidinotransferase (GATM), the rate limiting enzyme of creatine biosynthesis, in adipose tissue predisposed mice to dietinduced obesity [132]. In addition to the mitochondrially localized CKMT1 and CKMT2, creatine kinase B-type (CKB) can traffic to mitochondria [131]. Recent proteomics data suggest that CKB is the most abundant creatine kinase isozyme in brown and beige fat mitochondria. CKB is induced by cAMP signaling in mouse and human adipocytes and adipose-selective deletion of CKB reduces thermogenic capacity and predisposes to obesity. The identity of the phosphatase that catalyzes the dephosphorylation of PCr to creatine is not currently known.
Lipid cycling is a futile cycle that couples the intracellular hydrolysis of triglycerides in adipocytes and the re-esterification of fatty acids derived from intracellular or external sources. It has been observed that chronic β3-adrenergic activation simultaneously increases lipolysis and de novo lipogenesis in adipose tissues and the increased lipid turnover in this setting is associated with upregulation of genes involved in fatty acid synthesis, fatty acid oxidation, and glycerol metabolism [12,133]. ATGL, the rate limiting enzyme for lipolysis, is required for these effects. Induction of adipocyte glycerol kinase, a key enzyme that converts glycerol to glycerol-3-phosphate in an ATP-dependent reaction, allows the recycling of glycerol in triglyceride resynthesis. Because ATP is consumed in the re-esterification, simultaneous operation of lipolysis and lipogenesis is thought to act as an ATP sink and drive thermogenic respiration. Recent data suggest that futile lipid cycling is increased in BAT lacking UCP1 [134].
Another UCP1-independent thermogenic mechanism involving a futile cycle in adipocytes is ER Ca2+ cycling [11]. In this mechanism, ATP-consuming translocation of Ca2+ into the ER by sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) and the release of Ca2+ from the ER into the cytosol via the Ca2+ release channels constitute a potential futile cycle that enhances thermogenic respiration. Ca2+ cycling-driven thermogenesis has been described in skeletal muscle [135]. A small transmembrane protein called sarcolipin (Sln) uncouples SERCA ATPase activity from Ca2+ transport to create an ATP sink and serves as a regulator of muscle thermogenesis. Sln deleted mice develop hypothermia upon a cold challenge and are also predisposed to diet-induced obesity [136]. Beige fat has also been reported to utilize Ca2+ cycling in adrenergic receptor-stimulated thermogenesis. UCP1 null mice transgenically overexpressing Prdm16 in adipose tissue tolerate cold exposure and have increased expression of SERCA2b in inguinal WAT that may reflect higher levels of Ca2+ cycling-mediated thermogenesis [11]. SERCA2 levels are also induced in inguinal WAT by chronic cold exposure and in primary beige adipocytes by cAMP activation. Genetic disruption of SERCA2b impairs UCP1-independent thermogenesis in primary beige adipocytes. These observations raise the possibility that modulating Ca2+ cycling in beige fat could offer potential therapeutic approaches. Recent studies suggest that neuronatin (Nnat), an ER transmembrane protein whose expression is suppressed by thermogenic stimuli in WAT, impairs SERCA-mediated Ca2+ transport and heat dissipation and may serve as a potential target for regulating thermogenesis [137].
Human studies
Following the demonstration of thermogenic fat depots in adult humans, numerous studies utilizing fluorine 18-fluorodeoxyglucose-positron emission tomography-computed tomography (18F-FDG-PET-CT) imaging techniques have been performed in human subjects [6-9]. An inverse correlation between BAT activity and BMI was observed, raising the possibility that a higher BAT activity may protect from obesity [6,7]. BAT was less prevalent in older individuals and during the summer months, inversely correlating with ambient temperature [6,138]. A retrospective study of 134,529 18F-FDG-PET-CT scans has linked BAT to better cardiometabolic health, including lower odds of T2DM, dyslipidemia, coronary artery disease, cerebrovascular disease, congestive heart failure, and hypertension [138]. The presence of BAT correlated with better blood glucose, triglyceride, and high-density lipoprotein levels and the benefits of BAT were found to be greater in overweight and older people. Other studies have found an inverse correlation between BAT volume and hemoglobin A1c levels [139,140]. Several small experimental clinical studies have shown that activation of BAT by cold enhances increased insulin sensitivity, tissue fatty acid uptake, BAT lipid metabolism gene expression, and mobilization of lipids from the periphery [109,141-144]. While human BAT activity has generally been studied by 18F-FDG-PET/CT imaging in the setting of acute cold exposure, one study examined BAT substrate utilization under warm (25°C) and cold (17°C) conditions by combining microdialysis of blood samples from supraclavicular BAT and 133xenon washout and concluded that BAT has higher glucose uptake and lactate release compared with WAT [145]. Cold exposure selectively further increased blood flow and glucose uptake in BAT. Thus, even under warm conditions, human BAT is metabolically active and has the capacity for high glucose uptake, underlining its potential therapeutic utility.
As an alternative to subjecting individuals to prolonged cold exposure, pharmacological stimulation of β3-adrenergic signaling has been used to activate BAT thermogenesis and promote the browning or beiging of WAT. Previously studied β3-adrenergic agonists have been associated with unacceptable cardiovascular side effects limiting their use. The most attention has focused on mirabegron, a β3-adrenergic agonist that has already been approved by the U.S. FDA for treating overactive bladder. Treatment with doses greater than 150 mg per day produced negative cardiovascular effects but a dose of 100 mg per day for 4 days increased the energy expenditure without increasing blood pressure or heart rate [146]. A 4-week treatment of healthy female participants with 100 mg mirabegron daily increased BAT metabolic activity, whole-body resting energy expenditure, insulin sensitivity, and insulin secretion without changes in body weight or composition [147]. A 12-week treatment of obese, insulin-resistant men with 50 mg mirabegron per day improved glucose homeostasis and induced some thermogenic marker genes such as UCP1, transmembrane protein 26 (TMEM26), and cell death inducing DFFA like effector A (CIDEA) in subcutaneous WAT, but did not increase BAT volume based on PET-CT imaging [148]. It has been suggested that the FDA-approved dose of 50 mg daily is not sufficient to stimulate human BAT due to low β3-adrenergic receptor expression in human supraclavicular BAT and the effect seen with high doses of mirabegron are instead due to β2-adrenergic receptor agonism [149]. It is not known if β2-adrenergic receptor agonists are more efficacious than mirabegron in enhancing BAT activity and whether serious side effects occur due to β2-adrenergic receptor expression in smooth muscle cells in the respiratory tract, gastrointestinal tract, and skeletal muscle cells. However, another study has reported detecting significant β3-adrenergic receptor expression in primary adipocytes from human supraclavicular BAT and that the β3-adrenergic receptor is required for thermogenesis and lipolysis [150].
CONCLUSIONS
Experimental and clinical research over the past several decades have uncovered an enormous amount of knowledge about adipose tissue and its importance in metabolic health. There are now well-established links between adipose tissue and the regulation of systemic glucose and lipid metabolism, energy storage and expenditure, inflammation, and inter-tissue communication with other metabolic organs. Despite the remarkable progress that has been made, much remains to be elucidated about the detailed mechanisms through which adipose tissue dysfunction contributes to the pathogenesis of metabolic disorders. Because of the rising prevalence of obesity and associated comorbidities, there is a pressing need to develop effective therapeutics. Emerging areas of investigation include the use of exosomes to deliver miRNAs, pharmacological activation of BAT thermogenic activity, stimulation of peripheral lipid clearance by WAT, and administration or inhibition of specific adipokines or batokines.
 XML Download
XML Download