

 PDF
PDF Citation
Citation Print
Print


TO THE EDITOR: Hepatosplenic T cell lymphoma (HSTCL) is a rare subtype of T cell lymphoma that accounts for less than 3% of all peripheral T-cell lymphomas. It is prevalent in adolescents and young adults (median age ∼35 yr) and is derived from cytotoxic T cells, usually of γδ T cell receptor (TCR) type [1, 2].
HSTCL is characterized by a triad of cytopenia, B symptoms, and hepatosplenomegaly, usually without lymphadenopathy or peripheral lymphocytosis [3]. Furthermore, the disease progresses with a poor response to currently available therapies [4].
Among pediatric patients, the most commonly encountered non-Hodgkins’s lymphoma (NHL) are Burkitt lymphoma, diffuse large B-cell lymphoma, and T cell lymphoblastic lymphoma; other forms of NHL are fairly rare [5]. Here, we report one case of HSTCL from our institute, highlighting its unusual presentation and clinical and pathological features, with the utilization of flow cytometry and immunohistochemistry (IHC) to detect HSTCL.
CASE REPORT
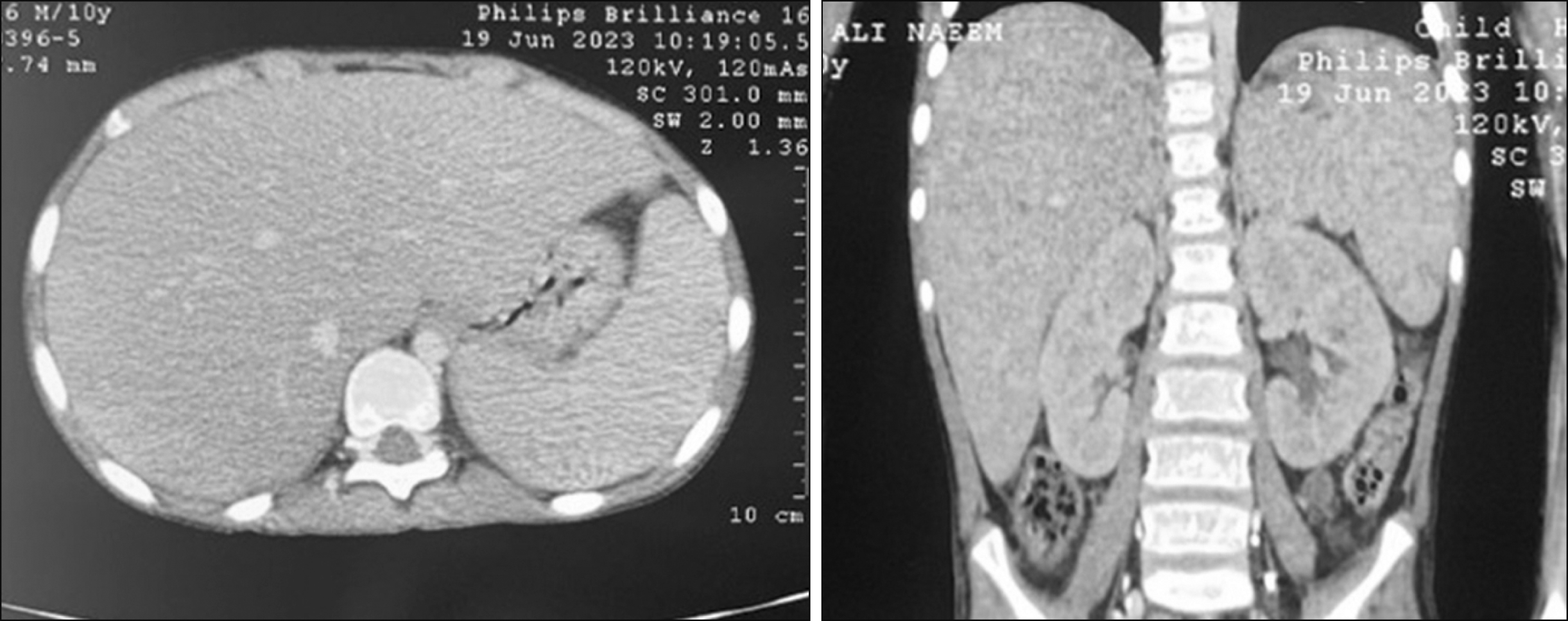
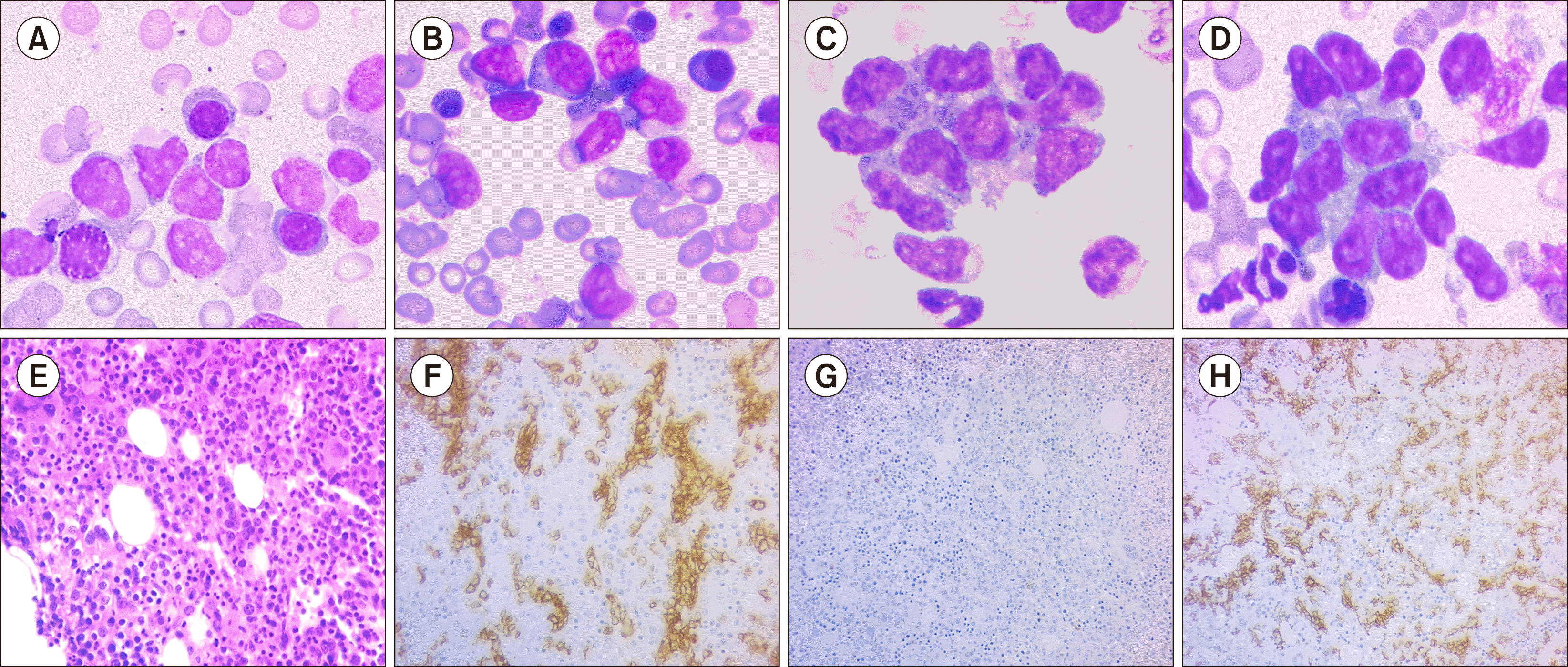
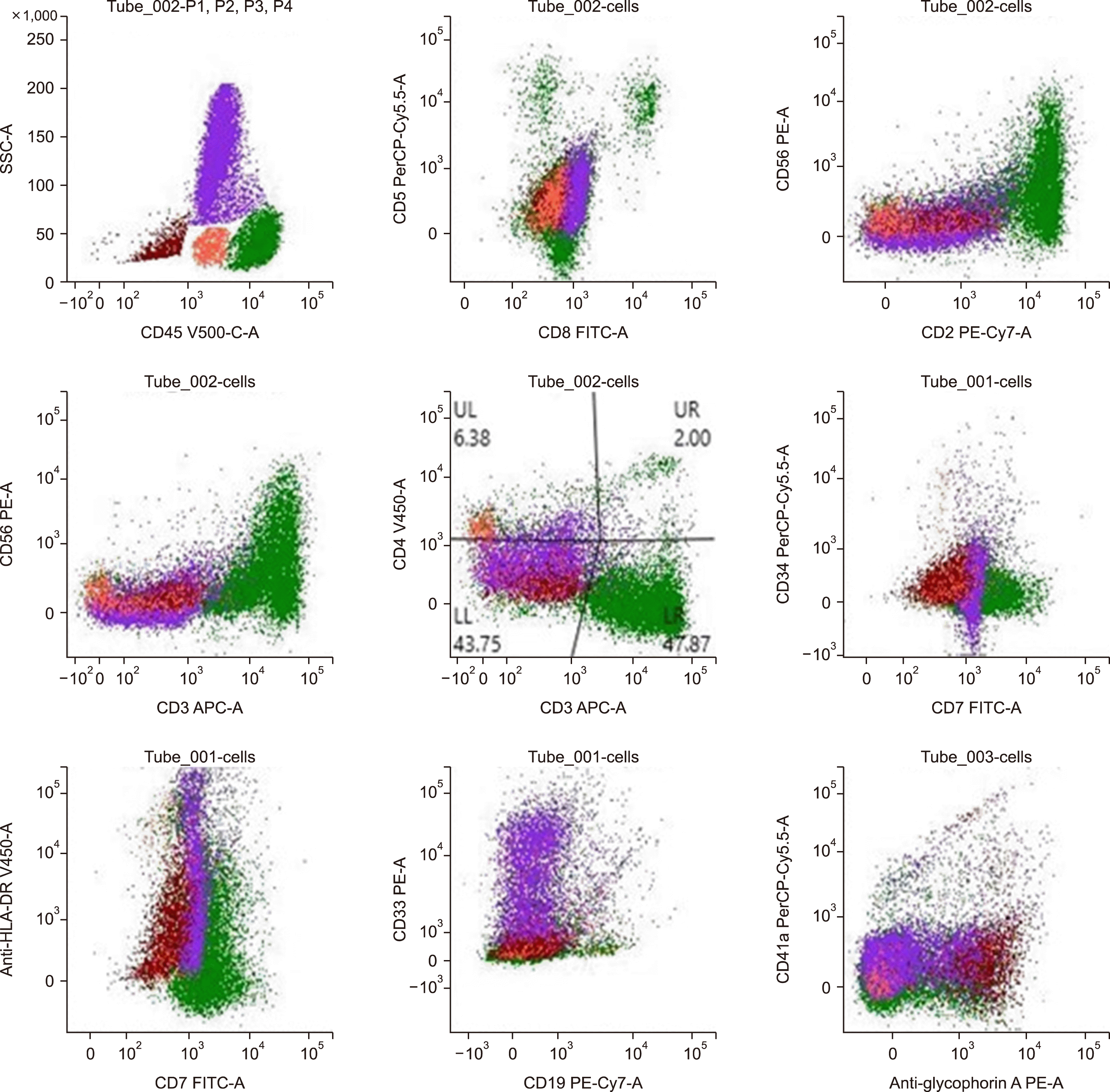
A 10-year-old male child who was immunocompetent and had no significant medical history presented himself with a history of intermittent high-grade fever, bone pain, and diffuse macular rash over the face, neck, and chest for 1 month before seeking medical attention. He was found to have marked hepatosplenomegaly without lymphadenopathy, and his initial blood work revealed mild normochromic anemia (10.0 g/dL, MCV: 84 fL), thrombocytopenia (87×109/L), and a total white blood cell count of 9.8×109/L (NEU: 5.2×109/L, LYM: 3.3×109/L). Rare atypical lymphoid cells with a suspicious morphology were initially observed (<1%) on blood film examination. Abdominal computed tomography (CT) revealed marked splenomegaly with a splenic size of 14.5 cm. His liver size also increased (19 cm) with no focal lesions in the spleen or liver (Fig. 1). Over the next few days, his platelet count dropped (66×109/L) with marked reticulocytosis (540×109/L). Although his hemoglobin level did not change, the direct Coombs test was negative, and his total serum bilirubin and lactate dehydrogenase measured 1 mg/dL and 1,440 U/L, respectively. The infection screen was unremarkable, with a negative EBV serology. Bone marrow (BM) aspirate smears revealed richly cellular marrow with tumor cells (43%) exhibiting somewhat blastoid morphology, small to medium size, pale basophilic cytoplasm, and minimally condensed chromatin with prominent nucleoli. Furthermore, many cells exhibited irregularities in their nuclear outline with cytoplasmic projections; these neoplastic cells were scattered among what appeared to be preserved hemopoietic elements as well as being arranged in cohesive clusters, raising the suspicion of acute leukemia vs. BM involvement by metastatic carcinoma (Fig. 2A–D). BM biopsy revealed subtle interstitial and intrasinusoidal infiltration by neoplastic cells, although this pattern was difficult to observe without the aid of IHC (Fig. 2E–H). The cerebrospinal fluid analysis yielded negative results. Flow cytometric analysis of the BM aspirate revealed the presence of approximately 48% abnormal T cells within the presumptive lymphocyte region (bright CD45 with low side scatter) expressing the following immunophenotypes: bright sCD3 and CD2 with dim CD7 and weak CD56 expression. The neoplastic cells were negative for cytoplasmic CD3, CD5, CD4, and CD8 (Fig. 3). The cells did not express immaturity markers (CD34, CD117, TdT) or B-lymphoid or myeloid markers. IHC CD3 staining of the BM biopsy specimen revealed a typical intrasinusoidal pattern of infiltration, and the lymphoma cells were positive for gamma- and negative for beta-IHC staining (Fig. 2G, H). Given the unique presentation, morphology, and immunophenotype, a diagnosis of HSTCL was made. The patient was treated according to the high-risk international Berlin-Frankfurt-Münster protocol. Induction was initiated using vincristine, daunorubicin, asparaginase, and steroids for 4 weeks; lymphoma cells constituted about 40% of marrow nucleated cells in the post-induction BM smear examination; thus, induction was extended for further 2 weeks using the same drugs, after which the patient achieved morphological remission with <5% malignant cells in the BM; however, the patient developed acute respiratory distress due to bronchopulmonary aspergillosis with many hemophagocytic figures in the BM, which can explain the persistent anemia and thrombocytopenia. 4 months post induction, the patient had relapsed with approximately 35% lymphoma cells in the BM examination, and the relapse protocol was initiated with intrathecal methotrexate, intravenous vincristine, and idarubicin with intramuscular asparaginase and oral dexamethasone for 4 weeks.
DISCUSSION
HSTCL is a rare subtype of NHL; since its discovery in 1990, there have been about 200 cases reported in the literature [6]. The pathogenesis of HSTCL is poorly understood; however, it is suggested that persistent antigenic stimulation may play a role [4]. The disease is most common in young adults and adolescents, with a male-to-female ratio of about 9:1 [4, 6]. About 20–30% of HSTCL cases are associated with chronic immunosuppressive disorder or the long-term use of immunosuppressive therapy [6, 7].
Patients present with marked hepatosplenomegaly; lymphadenopathy is unusual and has been reported in less than 25% of patients [1, 4]. Hemophagocytic syndrome has also been reported and can result in a rapid clinical course [6]. The patient in our study presented with persistent cytopenia despite rare lymphoma cells on post-induction BM smear examination, which can be explained by secondary hemophagocytic syndrome with hemophagocytosis of BM cells and elevated serum ferritin and triglyceride levels.
BM is often involved with peripheral thrombocytopenia or pancytopenia [1, 2, 8]. While the involvement of the BM is common, peripheral blood lymphocytosis is unusual at presentation, and only 1–2% of cases have reported it; though, blood involvement may be observed during advanced stages [1, 5, 6]. The spleen and liver show a predominant sinusoidal infiltration by monotonous neoplastic cells of medium size [1]. However, splenectomy is not essential, and an accurate diagnosis based on BM examination or liver biopsy with immunophenotyping should suffice [5, 9]. BM is the usual biopsy site for diagnosis. Positive biopsy results usually show intrasinusoidal lymphoid infiltration of cytotoxic T-cells and are difficult to identify without the aid of IHC [1, 4]. Interstitial and mixed sinusoidal and interstitial infiltration have also been reported [4], which was observed in the patient in our study. Generally, HSTCL cells are small to medium size with mature chromatin and irregular nuclear outline, inconspicuous nucleoli, and variable amounts of cytoplasm [6, 8]. Cytological atypia, in terms of large cells or blastoid morphology mimicking acute leukemia, has also been reported [1]. In our study, the cells were mostly of primitive morphology with minimally condensed chromatin and prominent nucleoli, which misled the initial diagnosis of acute leukemia; however, this possibility was ruled out by flow cytometry as the cells were CD45 bright, sCD3+, and negative for immaturity markers as well as the sinusoidal pattern of BM infiltration. Unusual morphological patterns mimicking metastatic marrow infiltration have also been reported [8], which was observed in our patient who exhibited lymphoma cells arranged in clusters; the possibility of BM involvement by metastatic cancer was also considered. The neoplastic cells are positive for surface CD3, CD2, and CD7 and are usually double negative (CD4-/CD8-), although CD8 can be expressed in some cases [6]. HSTCL cells are negative for CD5, TdT, and CD34, and most of the cases are CD56+ with expression of TCR gamma/delta [6]. The expression of αβ TCR has been reported in 20% of HSTCL cases, mainly in female patients and about 33% of patients over 50 years, and has been associated with worse prognosis [6, 9]. T-cell receptor β and λ genes are usually rearranged in HSTCL [6]. Most cases have Isochromosome 7 and trisomy 8 (63% and 50%, respectively), and about 40% have STAT5B and STAT3 mutations [1, 2, 6]. However, a genetic study was not performed for our patient.
Diagnosing HSTCL is challenging, and other lymphomas, such as splenic diffuse red pulp lymphoma and splenic marginal zone B-cell lymphoma, which show intrasinusoidal BM involvement, should be considered as a differential diagnosis [4]. The 5th World Health Organization classification of hematolymphoid neoplasms recognized four entities of γδ lymphomas: HSTCL, large granular lymphocytic leukemia (T-LGL), primary cutaneous gamma delta lymphoma (PCGDTL), and monomorphic epitheliotropic intestinal T-cell lymphoma (MEITL). Furthermore, all these lymphomas should be considered when a gamma delta type of T cell neoplasm is encountered during diagnosis [10]. The gamma/delta T-LGL has some features that may overlap with HSTCL, especially in cases where HSTCL shows CD8+ in flow cytometry. Other shared features include hepatosplenomegaly, sinusoidal marrow involvement, cytopenia, and a history of autoimmune diseases [4, 6]. In our patient, T-LGL was excluded due to the patient’s age, absence of large granular lymphocytes, and the immunophenotype (T-LGL cells are CD4-, CD8+, CD57+, and CD56-, and the majority of cases are positive of αβ TCR) [1]. PCGDTCL is an aggressive type of lymphoma that presents with skin plaques and ulcerations without splenic or BM involvement [10]. Despite our patient presenting with macular skin rash, this type of lymphoma was excluded based on the nature of the rash and the presence of BM, spleen, and liver involvement, although on reviewing the existing literature, HSTCL with skin involvement (proven by skin biopsy) has been reported twice. Other differential diagnoses of HSTCL include aggressive natural killer cell leukemia (typically negative for surface CD3 on flow cytometry) and T-lymphoblastic leukemia [dim expression of CD45 and surface CD3 with expression of immaturity markers (TdT/CD34/CD1a)] [4, 6, 8].
The disease has an aggressive course, and most cases will relapse. Moreover, there is no known optimal therapy [1, 2, 7]. With conventional chemotherapy, complete remission is uncommon, and most patients die within two years of diagnosis [5, 9]. Long-term remission can be achieved by allogeneic stem cell transplantation, and studies have suggested that both autologous and allogeneic transplants may confer a potential cure for the disease with an estimated 3 years overall survival after allogeneic transplantation of 56% [2, 8].
One major limitation regarding staging and assessing the extent of lymphoma is the unavailability of bone and PET scans for the patient at diagnosis, which makes assessment of response for chemotherapy and evaluation for achievement of remission challenging.
CONCLUSION
HSTCL is an aggressive extranodal type of T-cell lymphoma. Diagnosing HSTCL is challenging because it is uncommon and rarely encountered in clinical practice. To the best of our knowledge, this is the first pediatric case of HSTCL reported in Iraq, and it emphasizes the significance of considering the diagnosis in patients with splenomegaly and cytopenia without lymphadenopathy. Despite the misleading morphology often encountered during diagnosis, the typical immunophenotype and sinusoidal BM involvement can provide clues for an appropriate diagnosis.
 XML Download
XML Download