

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Rheumatoid arthritis (RA) is a systemic inflammatory autoimmune disease that preferentially affects small joints. Compared with the general population, patients with RA have an increased risk of premature death [1,2]. Cardiovascular disease (CVD), caused mainly by atherosclerosis, has the greatest effect on mortality in patients with RA [3]. Traditional risk factors such as age, dyslipidemia, hypertension, obesity, lack of exercise, and diabetes mellitus, as well as inflammation caused by RA itself and some medications that target inflammation, increase the risk of CVD [3].
The Janus kinase (JAK) family comprises receptor-associated tyrosine kinases that play roles in important biological processes by acting as signaling molecules downstream of type I and type II cytokine receptors [4]. Janus kinase inhibitors (JAKi) are targeted synthetic disease-modifying antirheumatic drugs (DMARDs) that are approved by the US Food and Drug Administration, the European Medicines Agency, and the Korean Ministry of Food and Drug Safety for the treatment of RA. However, the results of a recent postmarketing ORAL Surveillance study, which compared tofacitinib (a JAKi) with tumor necrosis factor-α inhibitor (TNFi) therapy in older patients with RA who have cardiovascular risk factors, revealed that tofacitinib failed to demonstrate noninferiority for major adverse cardiovascular events (MACE) [5].
According to the ‘lipid paradox in RA,’ patients with severe, untreated RA have lower cholesterol levels, but the risk of CVD is high [6]. However, although treatment of active RA can lead to elevated levels of total cholesterol (TC), low-density lipoprotein cholesterol (LDL-C), and high-density lipoprotein cholesterol (HDL-C), in conjunction with reduced levels of inflammation, the risk of CVD is low [6]. JAKi and TNFi effectively reduce inflammation and increase serum lipid levels [7-9]. However, the incidence of MACE is different [5]. One possible explanation may be the degree of cholesterol increase. In a clinical trial, JAKi-treated patients showed greater increases in LDL-C and HDL-C than those treated with adalimumab [10].
Slow accumulation of LDL and other apo B-containing lipoproteins in the arterial wall during young adulthood and middle age increase the total atherosclerotic burden [11]. Non-HDL-C includes all the cholesterol in all atherogenic lipoprotein particles, a combination of LDL-C, very low-density lipoprotein cholesterol (VLDL-C), VLDL remnants, and lipoprotein(a) [12,13]. LDL-C is the dominant form of atherogenic cholesterol, however, non-HDL-C is more atherogenic than either lipoprotein alone [12]. Many studies demonstrate that non-HDL-C can predict the risk of fatal or non-fatal ASCVD [13-15]. Moreover, fasting is not required for the calculation of non-HDL-C [16]. Surprisingly, there are few real-world data regarding changes in non-HDL-C in individuals taking biological or targeted synthetic DMARDs.
In this study, we focused on the association between lipid profile changes in patients with RA. To investigate whether JAKi increases cholesterol levels more than biological DMARDs, we compared changes in cholesterol between patients receiving different biological or targeted synthetic DMARDs (b/tsDMARDs). We also identified factors associated with increases in non-HDL-C after treatment with b/tsDMARDs.
MATERIALS AND METHODS
Patients
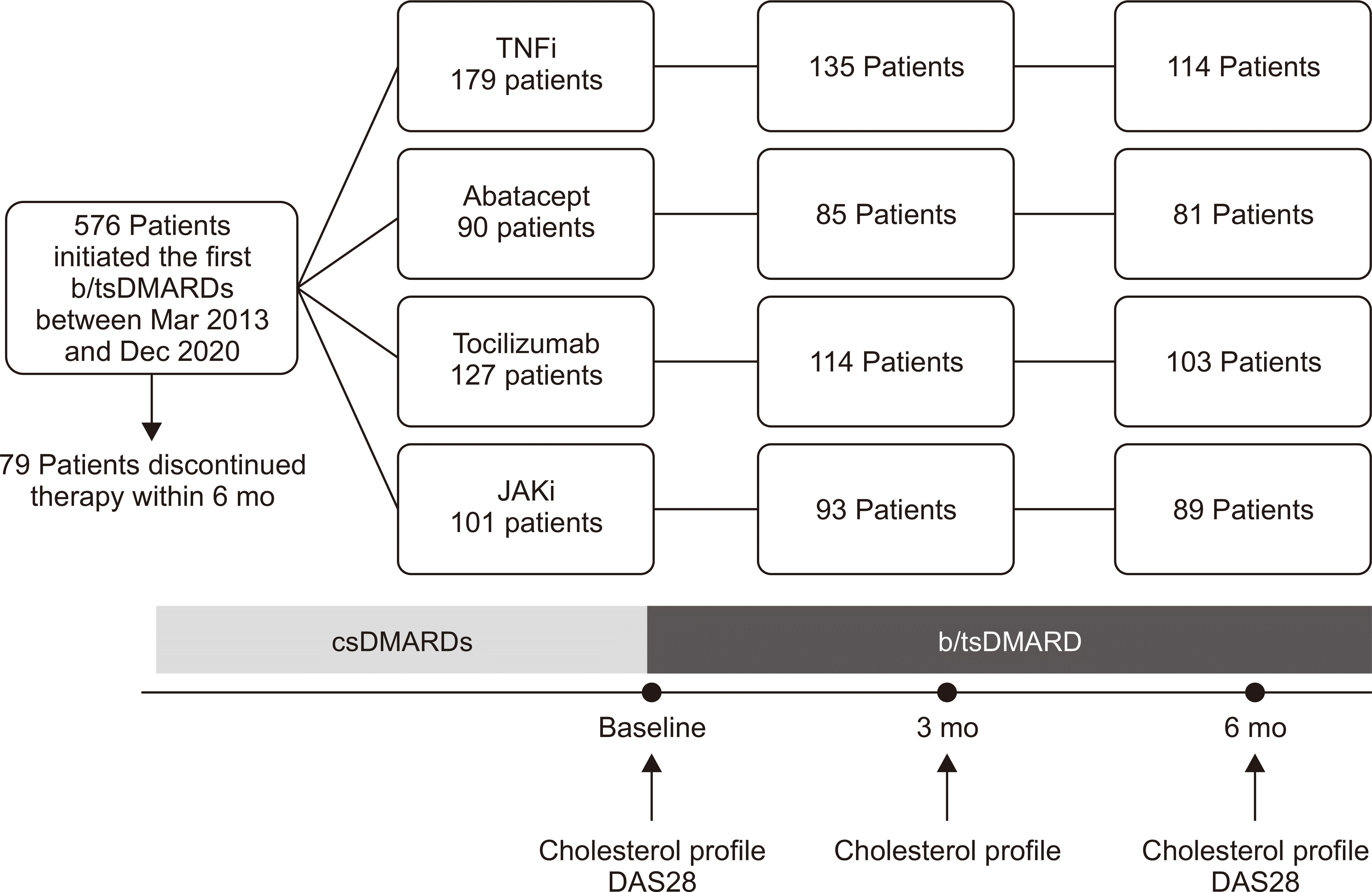
This retrospective study included patients treated at Seoul St. Mary’s Hospital. A total of 576 patients with RA began treatment with b/tsDMARDs between January 1, 2013 and December 31, 2020 (all were b/tsDMARDs-naïve prior to this). All patients were adults (≥20 years) who met the 2010 American College of Rheumatology (ACR)/European League Against Rheumatism (EULAR) classification criteria for RA [17]. All had active, moderate-to-severe RA (disease activity score in 28 joints [DAS28] >5.1) and had been treated for more than 6 months with two or more conventional synthetic DMARDs (csDMARDs), including methotrexate, unless they experienced adverse events or were contraindicated. These patients had follow-up assessments at 6 months, as required for continuation of national health insurance cover. Patients who did not have lipid profiles at 6 months were excluded from the analysis; 387 patients were finally included in this study (Figure 1).
In this study, patients received infliximab or its biosimilar agent, etanercept, adalimumab (a TNFi), tocilizumab (an interleukin [IL]-6 receptor inhibitor), abatacept (a T cell costimulatory blocker), or tofacitinib/baricitinib (a JAKi). Patients were classified into four treatment groups according to the mechanism of action of the b/tsDMARDs.
Ethical approval
This study was approved by the IRB of Seoul St. Mary’s Hospital (approval number: KC22RISI0953). Informed consent was waived for this retrospective study and patient information was anonymized and de-identified prior to analysis. The study was performed in accordance with the Declaration of Helsinki.
Variables
Data from the standard lipid profile (LDL-C, HDL-C, TC, and triglycerides) at baseline, 3 months (if available), and at 6 months follow-up were extracted from the hospital database. Non-HDL-C was calculated as non-HDL-C=TC−HDL-C. The level of LDL-C was measured in 89.7% of patients; and in patients who were not measured, LDL-C was calculated using the Friedewald equation, total cholesterol−HDL-C−(triglyceride/5). The measured and calculated values showed excellent reliability (intraclass coefficient, 0.855; 95% confidence interval [CI], 0.813~0.886: Cronbach's α=0.927).
The main demographic variables were age, sex, disease duration, comorbidities, presence of rheumatoid factor, and presence of anticyclic citrullinated peptide antibodies (ACPA). At the time of b/tsDMARDs initiation, RA disease activity was measured using the DAS28-erythrocyte sedimentation rate (ESR), and then again at the 6-month follow-up visit. Treatment response at 6 months was assessed using the DAS28-ESR and the EULAR response criteria [18]. Comedications, including non-steroidal anti-inflammatory drugs (NSAIDs), statins, glucocorticoids, and csDMARDs, were also reviewed.
There are no RA-specific recommendations for CVD risk prediction in Korea [19]. As the prevalence of CVD in patients with RA is increased to an extent that is comparable to that of patients with diabetes mellitus [3,4], RA was regarded as a borderline risk group. Hyper-LDL-cholesterolemia was defined as a serum LDL-C ≥130 mg/dL. Hypo-HDL-cholesterolemia was defined as a serum HDL-C level <40 mg/dL. Hypertriglyceridemia was defined as a serum triglyceride level ≥150 mg/dL. Dyslipidemia was then defined as satisfying one of the definitions stated above [19].
Some LDL-C levels were calculated, and fasting and hypertriglyceridemia could affect the result. However, the CVD risk associations of non-HDL-C are similar in the non-fasting and fasting population [20]. A significant increase in non-HDL-C was defined as ≥30%, as statins with moderate intensity are supposed to reduce LDL-C by 30% to 50% [12], and each statin induces equal reductions in LDL-C and non-HDL-C.
Statistical analysis
Demographic and disease characteristics are expressed as the mean±standard deviation or as a percentage (%). The levels of TC, LDL-C, HDL-C, triglycerides, and non-HDL-C are expressed as the mean percentage change from baseline; the treatment groups were compared using the analysis of variance (ANOVA), followed by post hoc analysis (the Tukey and Scheffe Method). The significance of the differences in DAS28, TC, LDL-C, HDL-C, triglycerides, and non-HDL-C between groups was analyzed using the Wilcoxon rank-sum test. The effects of clinical variables associated with a significant increase in non-HDL-C were estimated using binary logistic regression analyses. Model assumptions were checked using residual analysis. The results of these analyses are presented as odds ratios (ORs), with 95% CIs.
All p-values are two-sided, with p<0.05 considered statistically significant. SAS 9.4 (SAS Institute, Cary, NC, USA) software was used for data analysis, and graphs were drawn using GraphPad Prism 9.5 (GraphPad Software, San Diego, CA, USA).
RESULTS
Study characteristics
This study included 114 patients treated with TNFi, 81 with abatacept, 103 with tocilizumab, and 89 with JAKi (Figure 1). The baseline characteristics are listed in Table 1. Hypertension was less common comorbidities in the TNFi group than in the other groups, and type 2 diabetes mellitus was less common in the TNFi group than abatacept group.
Methotrexate in combination with abatacept was prescribed less frequently than methotrexate in combination with TNFi and JAKi (p<0.001 and 0.018, respectively). Similarly, methotrexate was prescribed less frequently for patients taking tocilizumab than for patients taking TNFi (p=0.04). Use of leflunomide in combination with TNFi was less common than in combination with abatacept and JAKi (p=0.006 and 0.038, respectively). Patients who started tocilizumab showed greater use of oral glucocorticoids than those who started JAKi (p=0.042).
Only 2.3% of patients did not respond to b/tsDMARDs at 6 months. There were more nonresponders in the group treated with TNFi than the group treated with JAKi (5.3% vs. 0%, respectively; p=0.028). The change in DAS28 was greater in patients treated with tocilizumab than in those treated with other agents.
Statins were prescribed to 55 patients (14.3%) prior to starting b/tsDMARDs; 17 patients began taking statins after starting b/tsDMARDs treatment.
Changes in the lipid profile
We found that TC, triglyceride, LDL-C, and non-HDL-C increased significantly in all of patients not taking statins (Table 2). There was no difference in lipid profile for each agent in TNFi and JAKi. The percentage change in TC and HDL-C showed a modest correlation with changes in the DAS28 (r= -0.141, p=0.014; and r=-0.138, p=0.016, respectively), and with changes in the CRP (r=-0.109, p<0.001 and r=-0.210, p<0.001). However, the percentage change in LDL-C and non-HDL-C did not correlate with changes in the DAS28 and CRP.
The percentage increase in TC was significantly higher in JAKi and tocilizumab users than in TNFi and abatacept users, respectively (Table 2). With respect to LDL-C and HDL-C, the increase in patients taking JAKi was greater than that in those taking TNFi and abatacept. In addition, the increase in non-HDL-C levels was higher in patients taking tocilizumab than that in those taking TNFi. The increase in HDL-C levels was higher in JAKi users than in TNFi and abatacept users. There was no significant difference between the treatment groups with respect to changes in triglycerides (Table 2).
At the 6-month follow-up, dyslipidemia occurred in 36.4% of patients. High non-HDL-C levels (≥160 mg/dL) were measured in 19.7% of patients, and high LDL-C levels (≥130 mg/dL) were measured in 23% of patients. Of the patients with dyslipidemia, only 15.6% started statin therapy.
Factors associated with a significant change in non-HDL-C
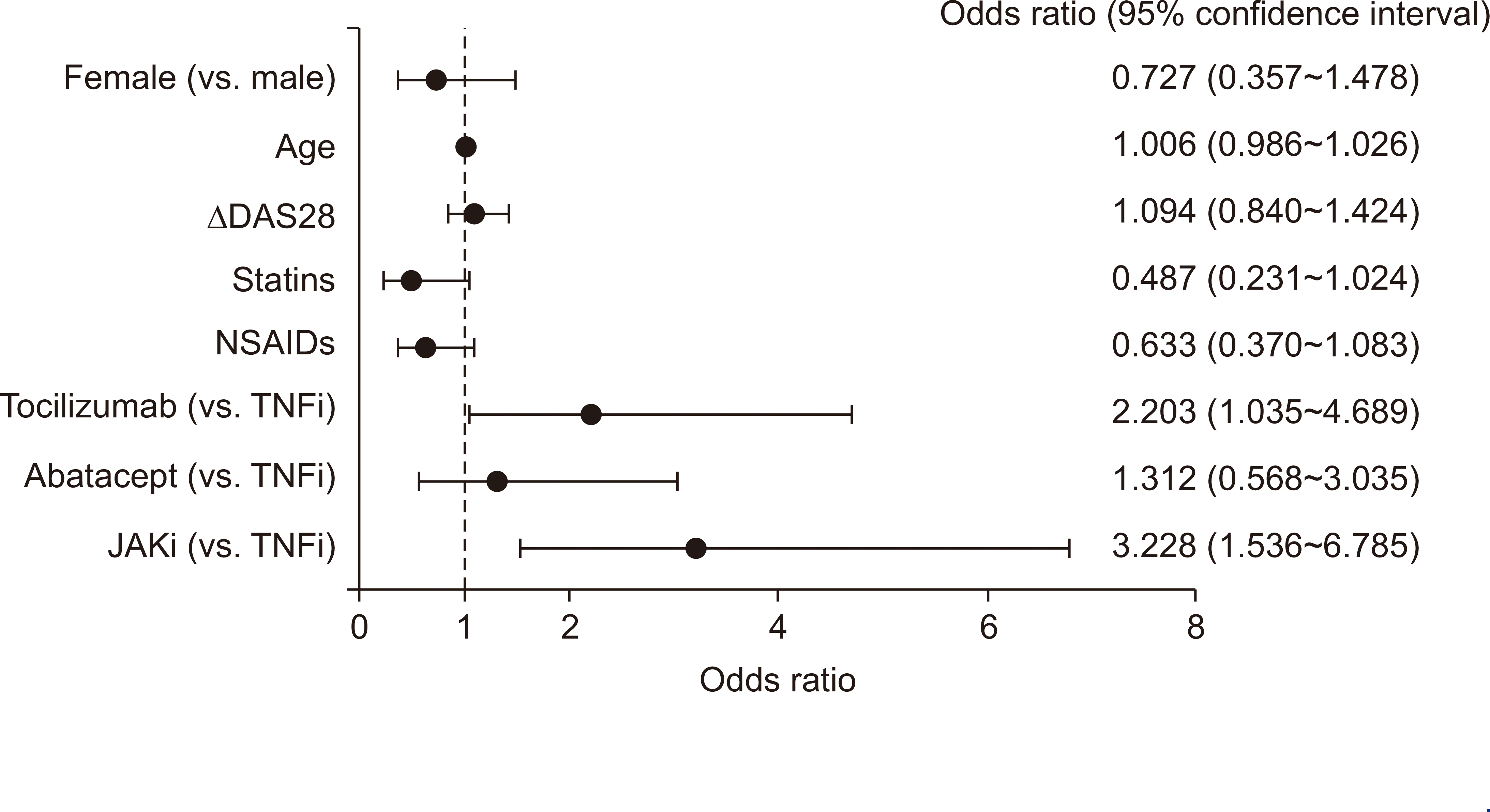
Multivariate analyses identified tocilizumab and JAKi as being associated with a significant increase in non-HDL-C when compared with TNFi (OR, 2.203; 95% CI, 1.035~4.689 for tocilizumab; OR, 3.228; 95% CI, 1.536~6.785 for JAKi). There was no association between changes in the DAS28, or combined use of csDMARDs, and increased non-HDL-C levels. However, concomitant use of statins was associated with a smaller increase (OR, 0.487; 95% CI, 0.231~1.024; p=0.058) (Figure 2).
DISCUSSION
As far as we know, this paper presents the first real-world data to simultaneously compare early changes in cholesterol profile in patients treated with one of the four clinically approved b/tsDMARDs as a first-line targeted therapy for RA. We demonstrate that JAKi and tocilizumab increase TC, LDL-C, non-HDL-C, and HDL-C levels to a greater extent than TNFi and abatacept.
Patients treated with tocilizumab and JAKi showed a significant increase in non-HDL-C when compared with patients treated with TNFi. This result is in agreement with those previously reported in randomized controlled trials [10,21]. TNF-α and IL-6 are inflammatory cytokines that have an atherogenic effect on endothelial cells [22,23]. Binding of TNF-α to its receptor triggers several signaling pathways, including nuclear factor κB (NF-κB), mitogen-activated protein kinases (MAPKs), and proteases [24]. IL-6 exerts its biological effects by binding to the IL-6 receptor, activating signal transduction via the Janus kinase-signal transducers and activators of the transcription factors (JAK-STAT) pathway and the Ras-MAPK pathway [25]. TNF-α and IL-6 receptor antagonists inhibit oxidized LDL-induced production of monocyte chemoattractant protein-1 (MCP-1), which plays a crucial role in the pathogenesis of inflammatory diseases and atherosclerosis [26,27]. However, blockade of each of these cytokines increases cholesterol levels by different degrees.
Here, we found that tocilizumab and JAKi resulted in a similar increase in cholesterol levels. Both agents affect the JAK-STAT pathway. Lipid metabolism is regulated by activation of the JAK-STAT signaling pathway in adipocytes [28]. IL-6 and JAKs are associated with STAT3, the most prominent transcription factor recruited to gp130 [29]. STAT3 promotes lipolysis and inhibits adipogenesis in mature adipocytes, as well as differentiation of preadipocytes [30], and activation of lipolysis in adipocytes increases HDL-C [31]. Therefore, inhibition of the STAT3 signaling pathway may block conversion of cholesterol to HDL, which is associated with an overall increase in non-HDL-C. Another study showed that cholesterol ester catabolism is higher in patients with RA, resulting in increased HDL-C, LDL-C, and TC [32]. Following tofacitinib treatment, the rate of cholesterol ester catabolism decreased, and cholesterol levels increased [32].
Abatacept modulates the costimulation of T lymphocytes. There are few data on the effects of abatacept on lipid profiles. A phase IIIb trial of abatacept combined with methotrexate versus adalimumab plus methotrexate revealed that those two combination therapies are associated with improvement in HDL function, despite increases in cholesterol levels [33]. A long-term observational study showed that abatacept and TNFi reduced the risk of CVD to a greater extent than csDMARD, whereas tocilizumab and tofacitinib did not [34]. Both TNFi and abatacept reduced inflammation while not increasing non-HDL-C levels significantly (compared with tocilizumab and JAKi), which may have helped to prevent CVD.
We found that in the real world, statins were prescribed to few patients. This may be due to passive control of high cholesterol levels following treatment of b/tsDMARDs, resulting from belief in the ‘lipid paradox in RA’ [35]. However, cholesterol is still the primary cause of atherosclerosis, which occurs in conjunction with inflammation [36]. Even in the young population, mildly abnormal lipid levels are associated with an increased future risk of atherosclerotic CVD events [37]. Our data also demonstrate that the level of TC and HDL-C increases as RA disease activity decreases. However, changes in non-HDL-C and LDL-C, which are crucial for atherogenesis [12,15], did not correlate with disease activity. Therefore, lipid-lowering therapy should be considered for RA patients showing a significant increase in cholesterol levels after treatment with b/tsDMARDs. Statins have beneficial effects beyond their lipid-lowering activities, including anti-inflammatory and proapoptosis effects on cultured RA synoviocytes [38]. Maintaining low LDL-C or non-HDL-C levels by treatment with statins may reduce the CVD risk in patients with RA.
This study has several limitations. First, atherosclerotic CVD risk estimation, which estimates the risk of a non-fatal or fatal CVD event in the next 10 years [12], was not evaluated. Some of the items for the risk estimation were missing, and these items were difficult to retrieve in retrospective studies. Second, the long-term changes in lipid profiles were not reviewed. This study focused on the early change of lipid profiles after initiating b/tsDMARDs, Lipid profiles are recommended to recheck 2 to 4 months after the inflammatory disease has been under control [12]. The long-term effect of b/tsDMARDs on lipid profiles has been studied in Korea which showed no difference between b/tsDMARDs [39]. Third, it was difficult to compare CVD events during follow-up because the observational period for JAKi was relatively short; this agent has been used as a first-line b/tsDMARDs only since 2017. To better elucidate the relationship between b/tsDMARDs, dyslipidemia, and CVD event in patients with RA, a prospective long-term follow-up study of patients in whom onset of RA occurred at a younger age would better identify the relative roles of inflammation and cholesterol in development of CVD.
CONCLUSION
Patients treated with b/tsDMARDs show increased levels of TC, LDL-C, and HDL-C. A significant increase in non-HDL-C was associated with the type of agent. Increased non-HDL-C levels were not associated with a reduction in the DAS28. Our data suggest that lipid profile should be regularly monitored in RA patients treated with b/tsDMARD and that prescription of statins should be considered for RA patients showing a significant increase in cholesterol levels.
 XML Download
XML Download