

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Aging is an unavoidable biological phenomenon, but the process can be delayed by a healthy lifestyle in humans. Some animal studies have shown that certain types of medication can delay aging [1], suggesting that aging may be categorized as an incurable but partially controllable disease [2]. Aging results in unhealthy conditions such as immune senescence, inflammaging, and a decline in cellular energy metabolism [3]. Centenarian studies and calorie restriction studies have consistently demonstrated that greater longevity can be achieved by reducing the risk of major age-associated diseases such as cardiovascular diseases, cancers, and neurocognitive disorders, suggesting that our lifespan and health span can be extended together [4].
Nutrition can affect gene expression in several ways including those occurring post-translation modifications such as protein phosphorylation, post-transcriptional modifications such as microRNA, or directly occupying receptors in DNA such as retinoic acid and vitamin D receptors. Epigenetics is the mechanism by which nutrition affects gene expression through transcriptional modifications. Epigenomics involves genome-wide epigenetic changes. Over the past several decades, the association between nutrition and DNA methylation—a major epigenetic phenomenon—has been extensively investigated, with particular focus on cancer and aging. The commonly observed pattern of age-associated DNA methylation is a decrease in global DNA methylation and increase in methylation of the promoters of critical genes [5], which is similar to that observed in cancer. However, the degree of DNA methylation-related changes in cancer is much wider and significant than those occurring during aging. Thus, aging, which is a major risk factor of cancer, can be hypothesized to provide an epigenetic milieu for the development of cancer [6]. DNA methylation has also been associated with DNA integrity. Deamination of methylated cytosine can induce C to T mutation, and this mutation rate is also correlated with advanced age [7].
An epigenetic diet was proposed in 2011 on the premise that many nutrients and bioactive food components can modify epigenetic phenomena such as DNA methylation and histone modifications, thereby promoting human health [8]. Many nutrients are involved in one-carbon metabolism, ultimately involving the synthesis of S-adenosylmethionine from methionine. S-adenosylmethionine is the unique methyl donor in the DNA methylation reaction. Thus, nutrients are expected to modify age-associated DNA methylation changes and reduce the development of age-associated disorders, ultimately extending the lifespan and health span [4].
In the present review, we focus on the relationship between one-carbon metabolism, DNA methylation, and aging to emphasize that the modulation of one-carbon metabolism with nutrition could be an epigenetic strategy to achieve healthy aging.
ONE-CARBON METABOLISM AND AGING
Multiple biological functions of one-carbon metabolism
One-carbon metabolism, which occurs in the cytoplasm, nucleus, and mitochondria, is a metabolic network that integrates major cellular functions with one-carbon nutrients, metabolites, and enzymes (Fig. 1) [9]. A pivotal role of one-carbon metabolism is the transmethylation pathway that produces S-adenosylmethionine and donates methyl moiety from S-adenosylmethionine to the DNA, proteins including histones, amino acids, lipids, neurotransmitters, and small molecules. Biological methylations are universal biochemical processes that occur in all the cells in the human body and controls gene expression, protein function, healing processes, cell energy, neurological function, liver detoxification, and immune function. Thus, biological methylations are critical for the maintenance of body functions. DNA methylation is a major type of biological methylation that has been investigated in embryonic development, aging, and cancer and is now known to be involved in the etiology of most major diseases such as cardiovascular diseases, neurocognitive diseases, metabolic disorders, endocrine diseases, and immune diseases [10].
Fig. 1
One-carbon metabolism comprising the transmethylation pathway, nucleotide synthesis pathway, transsulfuration pathway and remethylation pathway.
THF, tetrahydrofolate; DHF, dihydrofolate; SAdoMet, S-adenosylmethionine; SAdoHcy, S-adenosylhomocysteine; MTHFR, methylenetetrahydrofolate reductase; SHMT, serine hydroxymethyltransferase; TS, thymidylate synthase; DHFR, dihydrofolate reductase; MS, methionine synthase; BHMT, betaine-homocysteine methyltransferase; CBS, cystathionine β-synthase; MTHFD, methylene tetrahydrofolate dehydrogenase; MTHFS, methylenetetrahydrofolate synthetase; DNMT, DNA methyltransferase; TET, ten eleven translocation.
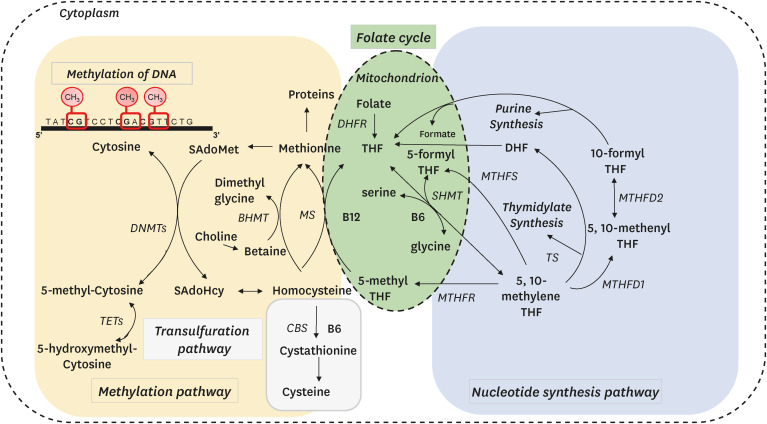
The second role is in the nucleotide synthesis pathway. Folate-mediated one-carbon metabolism produces thymidylate and purines for the synthesis and repair of DNA [9]. In this pathway, serine is the methyl donor, and glycine is the methyl acceptor. Hence serine and glycine act as methyl shuttles in a reversible reaction catalyzed by serine hydroxymethyltransferase. Inhibitors of the nucleotide synthesis pathway such as methotrexate and 5-fluorouracil have been used for cancer chemotherapy because they reduce the nucleotide pools that are needed for the growth of cancer cells.
The third role is the transsulfuration pathway, which produces cystathionine from homocysteine through a reaction catalyzed by cystathionine β-synthase and vitamin B6. Cystathionine is then hydrolyzed to cysteine, a precursor of taurine and glutathione, which have antioxidant properties and improve mitochondrial function [11].
The fourth role is the remethylation pathway, in which methionine synthase catalyzes the transfer of a methyl group from 5-methyltetrahydrofolate to homocysteine to form methionine. Vitamin B12 also acts as a cofactor in this reaction. Choline and betaine also catalyze the remethylation of homocysteine to methionine by betaine-homocysteine methyltransferase independent of folate and vitamin B12 [12].
Maintaining balance in one-carbon metabolism is critical because it integrates many biological processes such as methylation reactions, DNA synthesis and repair, antioxidant production, and homocysteine removal. Total plasma homocysteine level, S-adenosylmethionine and S-adenosylhomocysteine levels, and DNA methylation status have been used as indicators of one-carbon metabolism balance [1314]. Ultimately, several trials have been performed to maintain the balance of one-carbon metabolism using one-carbon nutrients [151617].
Aging and hyperhomocysteinemia
Evidence suggests that advancement in age tends to induce hyperhomocysteinemia [18], which is associated with geriatric multisystem problems including neurocognitive disorders, cardiovascular diseases, peripheral vascular disease, chronic renal disease, and sarcopenia [19]. Elevation of homocysteine level with aging is caused by reduced renal excretion of plasma homocysteine due to a decline in renal function, decreased intake of one-carbon nutrients, dysregulation of the methionine cycle, and decreased homocysteine remethylation and transsulfuration. Impaired homocysteine metabolism can lead to tissue degeneration through increased oxidative stress, inactivation of the nitric oxide synthase pathway, and mitochondrial dysfunction [20]. Interestingly, elevated homocysteine in the blood leads to its reentry into the cell and conversion into S-adenosylhomocysteine by reversing the reaction catalyzed by S-adenosylhomocysteine hydrolase, and possibly inhibiting methyl transfer to DNA [1421]. One-carbon nutrients such as folate, vitamin B6, and vitamin B12 can reduce plasma homocysteine levels; however, the results of clinical trials that have determined the effects of homocysteine reduction by B vitamins on the development of age-associated diseases have been inconsistent and contradictory [2223].
5-methyltetrahydrofolate and longevity
In a study involving Caenorhabditis elegans to determine the metabolic profiles of long-lived mutant models, folate metabolites demonstrated extensive quantitative changes in long-lived strains [24]. Among them, 5-methyltetrahydrofolate, the predominant form of circulating folate that serves as a methyl donor for the remethylation of homocysteine, showed substantially reduced levels, which subsequently reduced S-adenosylmethionine and increased homocysteine. More importantly, restoration of the folate pool through supplementation with 5-methyltetrahydrofolate reversed the longevity phenotypes, suggesting that reduced 5-methyltetrahydrofolate level and associated changes in one-carbon metabolism have a cause-and-effect relationship with longevity.
One-carbon metabolism and age-associated diseases
Over the past several decades, one-carbon metabolism has been investigated for its role in cancer development. Early animal studies demonstrated that a methyl donor-deficient diet, which is low in methionine, choline, folate, vitamin B6, and vitamin B12, can induce fatty liver changes and ultimately lead to liver fibrosis and cancer [25]. These nutrients used to be called lipotropes, which means compounds that prevent excess fat deposition.
Polymorphisms in genes encoding enzymes that mediate one-carbon metabolism are associated with an increased risk of cancer [2627]. These enzymes include methylenetetrahydrofolate reductase, serine hydroxymethyltransferase, methionine adenosyltransferase, methionine synthase, glycine N-methyltransferase, dihydrofolate reductase, thymidylate synthase, and cystathionine β-synthase. Some of these enzymes have been used as therapeutic targets for cancer chemotherapy; methotrexate inhibits dihydrofolate reductase and thymidylate synthase [28] and 5-fluorouracil inhibits thymidylate synthase [29].
Imbalance of one-carbon metabolism is highly associated with other age-associated diseases such as cardiovascular diseases [30] and neurocognitive disorders [31], whereas an adequate balance may have a preventive effect [32]. However, intervention studies have not clearly elucidated the preventive effects of one-carbon nutrient supplementation on the development of cardiovascular [3334] and neurocognitive diseases [35].
DNA METHYLATION AND AGING
DNA methylation is an epigenetic phenomenon that refers to the addition of a methyl (CH3) moiety to cytosine bases in the DNA. Similar to genetic phenomena, epigenetic phenotypes acquired through environmental factors can be inherited. However, in contrast to the genetic phenomena, DNA methylation does not affect DNA base sequences and can be modified by environmental factors including nutrition. Hydroxymethylation of cytosine at the same 5′ carbon site has been discovered [36] and suggested as a transient status of the active demethylation process of DNA methylation. Nowadays DNA hydroxymethylation is regarded to play a significant role in regulating gene expression [53738].
DNA methyltransferases (DNMTs) catalyze the transfer of methyl group from S-adenosylmethionine to cytosine at CpG residues in DNA to form 5-methylcytosine, whereas ten eleven translocation (TET) enzymes catalyze the transformation from 5-methylcytosine to 5-hydroxymethylcytosine and then 5-fomylcytosine, which ultimately convert to (unmodified) cytosine as a demethylation process. Age-dependent decreases in DNMT levels and low methyl donor nutrient or high homocysteine levels synergistically aggravate age-dependent DNA methylation changes and subsequently increase aberrant gene expression [39]. TET enzymes, which are critical for embryonic development via embryonic genome activation [40], can also be altered by aging [41]. In an animal study, old mice showed increased hepatic DNA hydroxymethylation compared with that in young mice, whereas no significant difference was observed in hepatic DNA methylation between the young and old mice [42]. Age-associated DNA hydroxymethylation in human peripheral blood cells is enriched in genes involved in immune processes, which may support a possible role in immunosenescence. In human T cells, the mRNA expression levels of TET1 and TET3 decrease with age, whereas that of TET2 is not influenced by age [43]. Further studies are needed to clarify the role of these demethylases in age-related DNA methylation and hydroxymethylation.
Conventional studies on DNA methylation and aging
Early epigenetic studies on aging have consistently demonstrated age-related global DNA hypomethylation and paradoxical site-specific DNA hypermethylation, which is associated with decreased expression of the corresponding genes. DNA methylation in senescent cells is enriched at gene-specific promoter sequences, particularly those that play important roles in senescence and cancer such as estrogen receptor alpha, insulin-like growth factor-II, p16, and E-cadherin [44]. In contrast, hypomethylation has been detected during aging in the CpG sites located in the gene body or intergenic area [45].
DNA methylation also becomes depleted with aging, particularly in constitutively heterochromatic repetitive DNA [44]. Histone proteins are integral for packing DNA in the form of chromatin; the compactness is determined by epigenetic phenomena, and it affects the expression of nearby genes. Chromatin, which forms the chromosomes, is of 2 different types; euchromatin and heterochromatin, which can be differentiated by the DNA methylation status. Euchromatin is less methylated, thereby less condensed and enriched with active genes, whereas heterochromatin is fully methylated, extremely condensed, gene-poor, and transcriptionally silent. Thus, age-associated DNA hypomethylation in the heterochromatin area results in heterochromatin loss. Heterochromatin domains established during early embryogenesis are reduced with age, causing global nuclear structural changes and aberrant gene expression [45]. These changes ultimately lead to cellular dysfunctions, exacerbated aging process, and possible tumor development.
Genome-wide DNA methylation and aging
Following the completion of the human genome project and the concurrent advancement of high-throughput technology, DNA methylation can be measured throughout the whole genome using next-generation sequencing after bisulfite modification of DNA. Unmethylated cytosine is deaminated to uracil using the bisulfite treatment; however, methylcytosine is resistant to deamination. This characteristic can differentiate cytosine and methylcytosine at the CpG sites. Whole genome-wide DNA methylation has also been determined using methylated DNA immunoprecipitation microarray methods that use chips containing known CpG dinucleotides and methylcytosine precipitation with antibodies against methylcytosine or methyl-binding proteins [46]. In general, genes associated with development are gradually hypermethylated with aging, which repress those genes, whereas genes involved in metabolism and protein ubiquitination are gradually hypomethylated with aging, which de-repress those genes [47].
A few historical studies have clearly demonstrated the genome-wide DNA methylation changes due to aging. These studies evaluated the methylation profile of DNA isolated from blood T cells, brain frontal cortex, and eye rod photoreceptors [48495051]. By nature, epigenetic changes are not the same among the cells, tissues, and organs. In fact, the epigenetic phenomenon is highly cell-specific and the effects of aging on DNA methylation differ among cells, even within the same individual.
Difference in DNA methylation between neonate and centenarian individuals
The difference in the genome-wide DNA methylation pattern of CD4+ T cells between a newborn and a centenarian was determined using whole-genome bisulfite sequencing and validated with a DNA methylation array using a commercially available microarray platform that can determine 450,000 preselected CpG-sites. Compared with that in the neonate DNA, centenarian DNA showed less methylated CpG sites in all genomic compartments including promoters and intra- and intergenic regions, and less association with neighboring CpGs. However, the centenarian DNA showed hypermethylation at CpG islands in promoters and hypomethylation at CpG-poor promoters in regulatory regions. These observations were reproduced using a DNA methylation microarray. Interestingly, the genome-wide DNA methylation patterns of middle-aged individuals were intermediate to that of newborn and nonagenarian/centenarian groups [48].
Brain aging and DNA methylation
The brain could be the most vulnerable organ to assault by aging, because the decline of neurocognitive function is one of the most self-aware symptoms experienced by an individual during normal aging. In a human study that investigates the epigenetic effect of age-associated decline in brain function, whole genome-wide bisulfite sequencing was performed on the DNA extracted from the frontal cortex of the brain of individuals representing a wide age-range [49]. The aging pattern found in blood DNA, illustrated by global hypomethylation along with promoter hypermethylation, was not observed in aged brains. In brain DNA, the CpG segments that showed extreme DNA methylation status at a young age were more prone to undergo changes regardless of the genomic segment. Interestingly, among the significant age-associated CpG segments, active intergenic enhancers exhibited the most dynamic variation in DNA methylation with age.
In brain DNA, the age-associated segments that exhibit gradual hypermethylation with aging were highly enriched with genes associated with developmental ontology terms pertaining to the brain, consistent with the notion that DNA methylation downregulates neurodevelopmental genes [52]. In contrast, age-associated segments that were gradually hypomethylated with aging housed genes involved in metabolism. Overall, these observations support the significance of epigenetic regulation in aged brains.
Vison, aging, and DNA methylation
A decline in vision is a part of natural aging. An animal study analyzed genome-wide DNA methylation using next-generation sequencing and mouse rod photoreceptors in 4 different age groups (3, 12, 18, and 24 mon) to determine age-associated DNA methylation changes that might be responsible for the decline in vision due to aging. In this study, age-associated differentially methylated regions correlated with altered gene expression responsible for reduced rod function. Differentially methylated regions have also been linked to reduced basal mitochondrial respiration and increased fatty acid dependency. Thus, changes in DNA methylation with aging are associated with an age-associated decline in vision [50].
DNA methylation age and epigenetic clock
Many studies have consistently demonstrated that aging may increase or decrease DNA methylation in an area-specific manner with no impact on DNA methylation in certain area due to aging. These results led to subsequent studies that introduce the concept of an epigenetic clock as an aging epigenotype that determines the age-specific DNA methylation changes and may represent the biomolecular age, so called DNA methylation age [53]. It appears that the DNA methylation age can be a highly accurate molecular aging marker that may be useful in determining the biological age, longevity, and effects of anti-aging interventions [54].
Several aging studies have focused on DNA methylation age and the biological clock using DNA methylation array method on the premise that epigenetic deregulation is a hallmark of aging, and age-specific DNA methylation changes can be an epigenetic marker for aging [55]. Early on, 2 large-scale studies by Horvath [56] and Hannum et al. [57] characterized 353 and 71 CpG segments, respectively, which were differentially methylated during aging and utilized for DNA methylation age calculation. The main inference is that this first-generation DNA methylation age is well correlated with the chronological age and is expected to be a better aging marker than other biological aging markers such as telomere length [5859].
In contrast to the first-generation DNA methylation age, the second-generation DNA methylation age was developed in conjunction with other aging phenotypes. The DNA methylation PhenoAge was built using 513 CpG segments and 9 clinical variables associated with aging including C-reactive protein, albumin, glucose, creatinine, white blood cell count, lymphocyte proportion among white blood cells, mean red blood cell volume, red cell distribution width, and alkaline phosphatase. Thus, DNA methylation PhenoAge is useful for predicting age-associated conditions such as morbidity and mortality risks among individuals of the same chronological age [60].
The third-generation DNA methylation age is the DNA methylation GrimAge, which is computed using 1030 unique CpG segments, smoking pack-years, and 7 plasma protein markers (cystatin C, adrenomedullin, beta-2-microglobulim, growth differentiation factor-15, plasminogen activator inhibitor-1, leptin, and tissue inhibitor metalloproteinases 1), which are associated with morbidity or mortality. Thus, DNA methylation GrimAge can predict all-cause mortality and associations with lifestyle factors and clinical markers [61].
The 3 DNA methylation ages were determined in 715 participants of the VA Normative Age Study using a human DNA methylation microarray with bead chips and measuring the levels of 4 one-carbon nutrients (folate, vitamin B6, vitamin B12, and homocysteine). After adjusting for chronological age and lifestyle factors, this study demonstrated that plasma vitamin B6 levels were associated with decreased PhenoAge, whereas plasma folate levels were associated with increased PhenoAge and GrimAge. Furthermore, the cumulative level of the 4-metabolite mixture was associated with decreased GrimAge. This study may provide a clue regarding the influence of one-carbon metabolism on DNA methylation age markers [62].
A randomized controlled clinical trial demonstrated that healthy lifestyles with dietary supplementation could reduce the Horvath DNA methylation age. Interestingly, dietary supplementation significantly increased the level of 5-methyltetrahydrofolate, suggesting that the DNA methylation age can be lowered using one-carbon nutrients [63].
DNA methylation ages can be novel markers of biomolecular age calculated using the DNA methylation status of age-specific CpG dinucleotides [54]. DNA methylation ages may predict biological age, disease risk, and mortality risk, and may be modified using one-carbon nutrients. Collectively, a comprehensive evaluation of DNA methylation could be helpful in understanding the role of DNA methylation in aging and DNA methylation status could be a useful biomarker of aging and preventive target for age-associated disorders [6].
ONE-CARBON METABOLISM, DNA METHYLATION AND AGING
Early life nutrition affects later life health through DNA methylation
Early life nutrition is well known to affect later life health, and DNA methylation could be one of the underlying mechanisms. Early on, the famous Agouti mouse model was used to demonstrate that the levels of methyl contents such as folate, vitamin B12, methionine, and choline in the maternal diet changed the coat color in their offspring and impacted the later life obesity status and metabolic phenotypes through the altered expression of critical genes mediated by DNA methylation [64]. Epigenetic dysregulation commonly occurs with age; however, whether it is genetically determined (programed) or randomly induced by environmental influences has not yet been clearly elucidated. Nevertheless, evidence strongly suggests that early life nutrition may cause lifelong alterations in DNA methylation, which could be linked to age-associated disorders and longevity [65].
During embryonic development, DNA methylation plays a key role in guiding the differentiation process towards future lineages and preventing de-differentiation [66]. In fact, epigenetic patterns of DNA in the sperm and egg are delivered to the conceptus. However, during the very early development phase, genome-wide epigenetic reprogramming erases epigenetic marks originating from the sperm and egg and re-establishes its own epigenetic profile, including DNA methylation. These changes contribute to normal embryonic development and establish cell specificity. This reprogramming can be affected by one-carbon metabolism because epigenetic reprogramming that occurs in pluripotent stem cells requires an optimal S-adenosylmethionine pool for appropriate methyl delivery to maintain the pluripotent state [67]. Inadequate one-carbon metabolism can induce aberrant DNA methylation status and subsequently altered the epigenetic patterns in the gametes and early embryos, which may contribute to the development of future metabolic disorders and adult-onset diseases [68].
DNA methylation also regulates genomic imprinting, which is the monoallelic repression of a minor group of genes by DNA methylation [69]. The differentially methylated regions depending on whether they are inherited from the mother or father may be highly susceptible to changes in diet during the epigenetic reprogramming period, even though imprinting is hardly reversed during other life stages. More than 150 imprinted genes exist in the human genome and loss of imprinting is associated with the development of human diseases. The imprinted status affected by early life nutrition may influence the offspring’s later life health [70].
Changes in one-carbon metabolism and DNA methylation by one-carbon nutrients, other nutrients, and bioactive food components
As shown in Fig. 2 DNA methylation can be influenced by one-carbon nutrients because one-carbon metabolism regulates DNA methylation through the production of S-adenosylmethionine, the unique methyl donor, and S-adenosylhomocysteine, a methyltransferase inhibitor. Previous studies have demonstrated that diets deficient in multiple one-carbon nutrients can induce hepatic DNA hypomethylation [10]. However, a single one-carbon nutrient hardly affects DNA methylation status because nutrients and metabolites with similar functions work together in one-carbon metabolism. A human study that administered a food-frequency questionnaire to measure the dietary intake of one-carbon nutrients in adulthood suggested that it had little association with blood DNA methylation, and a single nutrient cannot induce significant global DNA methylation changes [71].
Fig. 2
A candidate mechanism for the axis of one-carbon metabolism, DNA methylation, and aging.
MTHFR, methylenetetrahydrofolate reductase.

Other nutrients and bioactive food components such as retinoic acid, selenium, resveratrol, curcumin, sulforaphane, and tea polyphenols could also modulate DNA methylation by altering the pathways in one-carbon metabolism or competitively inhibiting the DNMTs that catalyze the DNA methylation reaction.
Retinoic acid induces glycine N-methyltransferase, which belongs to the methyltransferase enzyme family and catalyzes the conversion of S-adenosylmethionine and glycine to S-adenosylhomocysteine and N-methylglycine [72]. Selenium increases the expression of serine hydroxymethyltransferase, which converts serine and tetrahydrofolate to glycine and 5-methyltetrahydrofolate, thereby increasing hepatic DNA methylation [73]. Selenium is also closely linked to the transsulfuration pathway [74].
Flavonoids, a group of phenolic substances in fruits and vegetables, and epigallocatechin-3-gallate (EGCG), a green tea polyphenol, can reduce global DNA methylation levels by inhibiting the activities of DNMTs. EGCG directly inhibits DNMT catalytic site. Sulforaphane, which is found in broccoli, may downregulate DNMT expression [75]. Curcumin from turmeric can also inhibits DNMTs [76]. Bioactive food components can inhibit the activities of DNMTs by increasing intracellular S-adenosylhomocysteine levels via catechol-O-methyltransferase enzyme reactions that uses S-adenosylmethionine as a cofactor. Resveratrol may modulate DNA methylation by altering the levels of S-adenosylmethionine and S-adenosylhomocysteine or by affecting the enzymes that catalyze DNA methylation [77].
Collectively, these nutrients and bioactive food components have been postulated to influence the aging process by modulating the DNA methylation profile (Fig. 2) [4]. An epigenetic diet that is rich in these nutrients and bioactive food components may mitigate the aging process through this mechanism [8].
Single nucleotide polymorphisms in one-carbon metabolism genes and DNA methylation
An intervention study found that supplementation with folate and vitamin B12 increased the global DNA methylation levels in individuals with the wild type methylenetetrahydrofolate reductase 677CC genotype, but not in those with the variant 677TT genotype with reduced enzyme activity. This effect was more prominent in the CpG islands and shores, which are characteristically CpG-dense regions, suggesting that this DNA methylation change may influence gene expression [78]. Methylenetetrahydrofolate reductase is a critical enzyme in one-carbon metabolism that determines the proportions of 5-methyltetrahydrofolate (folate for transmethylation pathway) and 5, 10-methylenetetrahydrofolate (folate for nucleotide synthesis). The common 677 C to T polymorphism in this gene affects the DNA methylation status and colon cancer development [79]. Interestingly, this study investigated the effects of dietary supplementation with folate and vitamin B12 on the reduction of DNA methylation age, as estimated using Horvath’s model. This effect was found only in women with the 677CC genotype [78].
Calorie restriction diet, one-carbon metabolism, and DNA methylation
In a yeast study, calorie restriction was found to significantly reduce the intracellular concentrations of S-adenosylmethionine, methionine, and cysteine as determined using liquid chromatography-tandem mass spectrometry [80], suggesting that calorie restriction has a significant impact on one-carbon metabolism.
Early studies on calorie restriction and DNA methylation focused on comparing DNA methylation patterns between young and old rodents fed either a calorie restriction diet or a control diet. In a short-term calorie restriction diet study [81] using young and old male specific pathogen free Sprague-Dawley rats and methylated DNA immunoprecipitation sequencing, both the old control rats and calorie restricted old rats showed hypomethylation in the CpG islands and repetitive regions, whereas the 5'-untranslated region (UTR), exon, and 3'-UTR were hypermethylated compared with that in the young rats. In contrast, the methylation status in the promoter and intron regions was decreased in the old control rats but increased in calorie restricted old rats. Pathway analysis revealed that hypermethylated promoters in the DNA from the old rats were associated with degenerative phenotypes such as cancer and diabetes mellitus, whereas the hypomethylated promoters in old rats were associated with the chemokine signaling pathway. However, the pathways enriched in old control rats disappeared from the calorie restricted old rats, suggesting that prevention of age-associated aberrant methylation by calorie restriction diet might be one of the underlying mechanisms by which calorie restriction extends lifespan and health span.
In another animal study, the influence of calorie restriction on age-associated changes in the DNA methylome was investigated using the blood DNA of C57BL/6 male mice from 16 different age groups (3–35 mon of age) [82]. Most of the methylome changes were found in late life (86% sites) and early life (14% sites), whereas few changes were observed in the middle-aged mice, suggesting that the primary changes during aging are not linear but occur mostly in late-life. In the early phase, calorie restriction shifted the DNA methylome in the same direction as normal aging, but the cumulative change during calorie restriction affected the methylation pattern differently compared with that during normal aging. This cumulative trend seemed to retain the young state by repressing age-associated methylome changes.
Both studies have suggested that calorie restriction may retard age-associated DNA methylation changes and maintain the young pattern, consequently reducing the development of age-associated diseases in calorie restricted animals. Interestingly, a most recent study demonstrated that aged mammalian tissues remember their young DNA methylation pattern, which can be accessed to restore the tissue function declined by aging [83].
Methionine restriction diet, one-carbon metabolism, and DNA methylation
The role of methionine restriction in healthy aging is similar to that of other restrictions such as calorie restriction, carbohydrate restriction, and time restriction. Methionine is an essential sulfur-containing amino acid that serves as a methyl donor in one-carbon metabolism and is involved in the synthesis of S-adenosylmethionine, cysteine, glutathione, taurine, and polyamines, which play a potential role in aging [84]. Methionine is a building block of proteins that also plays a critical role in initiating the process of assembling proteins. As methionine produces molecules that are essential for proper cell function, methionine is important for neonates and growing children. However, for elderly individuals limiting methionine intake is recommended to reduce the risk of age-associated disease such as cardiovascular disease [8586] and cancer [87].
Over the past decades methionine restriction has been associated with extended longevity and metabolic health [88]. Methionine restriction is also known to reduce the risk and overall mortality of cancer [8990]. Despite well-documented evidence for lifespan extension by methionine restriction, the underlying mechanisms have not been fully elucidated [9192939495]. Altered S-adenosylmethionine and S-adenosylhomocysteine pools due to methionine restriction and subsequent DNA methylation changes have been suggested as candidate mechanisms but the evidence is still controversial.
Interestingly, an S-adenosylhomocysteine supplementation study may provide a clue to understanding the mechanism underlying methionine restriction associated with healthy aging because methionine restriction alters the pool of S-adenosylhomocysteine [93]. A study involving yeast has suggested that S-adenosylhomocysteine may extend the lifespan in a manner similar to that of methionine restriction. In this study, S-adenosylhomocysteine supplementation led to inhibition of the mammalian target of rapamycin complex 1 (mTORC1), and the same result was observed following methionine restriction [92]. mTORC1 regulates cell growth, proliferation, metabolism, protein synthesis, and autophagy [96]. Furthermore, S-adenosylhomocysteine treatment activates adenosine monophosphate-activated protein kinase, a central regulator of energy homeostasis [97]. However, the precise mechanism underlying methionine restriction-mediated longevity needs to be elucidated.
The potential toxic effect of one-carbon metabolism and DNA methylation
Homocysteine is a non-protein-forming sulfur-containing amino acid produced from methionine known to induce cytotoxicity, inflammation, protein aggregation, and apoptosis. Additionally, a high concentration of homocysteine in the blood is known to damage the lining of the arteries and induce thromboembolism [98].
As homocysteine is cytotoxic, homocysteine formed in the cells is immediately released into the plasma, subsequently excreted through urine. Homocysteine in cells is also removed through the remethylation or transsulfuration pathways. Hyperhomocysteinemia due to the inherited deficiency of cystathionine β-synthase, decreased renal excretion due to impaired renal function, or imbalance of one-carbon metabolism has been well demonstrated to be associated with an increased risk of cardiovascular diseases [99] and neurocognitive disorders [100]. In fact, blood homocysteine levels tend to increase with age, which may be associated with an age-dependent decline in global DNA methylation [18]. Thus, hyperhomocysteinemia-mediated DNA hypomethylation may be the mechanism by which age-associated hyperhomocysteinemia increases the risk of age-associated diseases. The cause-and-effect relationship needs to be clarified.
Formaldehyde is a chemical compound generated through one-carbon metabolism. Despite being highly toxic they are utilized to form formate, which is a useful one-carbon metabolite [101]. Formaldehyde is a known carcinogen and teratogen that forms protein and DNA adducts and can lead to severe genotoxic stress [102]. Tetrahydrofolate is a methyl acceptor form of unsubstituted folate and is susceptible to oxidative breakdown, leading to the release of formaldehyde [103104]. Epigenetic demethylation can also release formaldehyde, which is a potential threat to genome integrity [105]. Once formed, formaldehyde reacts with glutathione and is ultimately converted to formate, which is then free to enter the one-carbon metabolism [102]. Formate is a source of 10-formyl-tetrahydrofolate and other one-carbon intermediates that are primarily used for purine and thymidylate synthesis. The purpose of mitochondrial one-carbon metabolism is to generate formate to be utilized in the one-carbon metabolism in the cytoplasm and nucleus [106]. In this regard, attention needs to be paid to the folate–formaldehyde–formate cycle, especially to the potential toxic influence.
CONCLUSION AND PERSPECTIVES
One-carbon metabolism is located at the center of metabolic networks, thereby mediating many essential physiologic processes such as DNA synthesis and repair, amino acid homeostasis, epigenetic maintenance, lipid metabolism, and redox defense. An imbalance in the one-carbon metabolism is associated with many pathologic processes including those leading to cancer, cardiovascular disease, neurocognitive disease, and fatty liver diseases. However, this aspect of one-carbon metabolism has not been as intensely investigated as its importance to human health. The status of one-carbon metabolism progressively changes with age and is influenced by diet. One-carbon metabolism conveys health effects through epigenetic mechanisms, especially DNA methylation, which is the most extensively studied epigenetic phenomenon.
Aging is accompanied by substantial changes in DNA methylation throughout the genome. A most recent study suggested that a loss of epigenetic information is a reversible cause of aging [107]. Thus, the modulation of one-carbon metabolism by nutrition may mitigate age-associated DNA methylation changes, which may delay the development of disorders or diseases related to age-associated DNA methylation changes. Aging cannot be prevented; however, age-associated DNA methylation changes that might cause age-associated disorders may be prevented or delayed.
In future, studies need to be performed to determine the ideal DNA methylation profile that can promote healthy aging. The means to achieving the ideal DNA methylation status also needs to be explored, especially through the modulation of one-carbon metabolism.
 XML Download
XML Download