

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
Disulfiram (DSF) is an Food and Drug Administration-approved anti-alcoholism drug that has been in use for over 60 years. As an inhibitor of aldehyde dehydrogenase, DSF leads to the accumulation of acetaldehyde and induces unpleasant feelings after alcohol consumption [1]. Recently, DSF has been highlighted as a potential anticancer agent for a wide range of cancers, and several clinical trials are ongoing [2,3]. DSF and its metabolites are known to exert anticancer effects through the aggregation of NPL4, a subunit of the p97/VCP segregase [4], inhibition of FROUNT, a chemokine signal regulator [5], induction of reactive oxygen species, and inhibition of NFκB [6,7]. Research on DSF-based cancer therapy has become increasingly active, as it has been revealed that the combination of DSF and copper enhances anticancer activity [8].
Dendritic cells (DCs) are professional antigen-presenting cells that are considered key immune cells in the initiation of antigen-specific immunity and tolerance [9]. DCs recognize danger signals from invading microbes and injured host cells, and these signals induce DC maturation. After maturation, DCs enhance the expression of co-stimulatory molecules and the production of cytokines, and then prime naïve T cells to activate adaptive immunity [10]. In autoimmune diseases, disruption of DC tolerance can promote autoreactive T cell responses and pathology. Therefore, the induction of DCs with tolerogenic properties has received attention for autoimmune treatment [11]. Moreover, DCs are critical in cancer treatment and play a role in determining the therapeutic efficacy of various cancer therapies, such as chemotherapy, adoptive T-cell therapy, and immune-checkpoint therapy. Many cancer immunotherapies target DCs, including tumor-derived antigen delivery, functional improvements of DCs, and DC vaccines [12].
Although DSF has been studied as an anticancer agent and DCs have an important role in immunity, the effect of DSF on DCs has not yet been investigated. Therefore, in this study, we assessed the cellular toxicity of DSF on DCs and the effect of DSF on the function of DCs from maturity to priming of naïve T cells.
Go to : 
METHODS
Animals and reagents
C57BL/6 and BALB/c mice were purchased from Samtako and housed in the animal facility of Jeju National University. In this study, 7–12-week-old mice were used in the experiments. Animal experiments in this study were performed in accordance with the Institutional Guidelines for Animal Use and Care of Jeju National University (2021-0047). DSF was purchased from Sigma-Aldrich and dissolved in dimethyl sulfoxide (Sigma-Aldrich). Lipopolysaccharide (LPS) was purchased from Sigma-Aldrich and dissolved in sterile phosphate-buffered saline. Doxorubicin (DOX) was purchased from TEVA-HANDOK and was used as a positive control.
Preparation of bone marrow-derived DCs
Bone marrow cells (BMs) were harvested from the femur and tibia of the C57BL/6 mice. To remove red blood cells, BMs were treated with an ammonium-chloride-potassium lysis buffer (Thermo Fisher Scientific). After filtration through a 70 µm cell strainer, the cells were cultured in 6-well plates in RPMI 1640 medium containing 5% fetal bovine serum, 100 U/ml penicillin-streptomycin, 2 mM L-glutamine, 10 ng/ml recombinant mouse granulocyte macrophage-colony stimulating factor (GM-CSF; PeproTech), and recombinant mouse interleukin (IL)-4 (PeproTech). The culture medium was replaced with fresh medium every two days. On the 6th or 8th day of culture, the floating cells were harvested and used as DCs.
Measurement of the metabolic activity and viability
To measure the metabolic activity of the cells, DCs, BMs, and spleen cells were treated with DSF and LPS and cultured in 96-well plates for 3 days. Each cell type was cultured at concentrations of 2 × 105, 5 × 105, and 2 × 106 cells/ml, respectively. For DCs and BMs, a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenylltetrazolium bromide (MTT) solution was added at a concentration of 0.5 mg/ml and incubated at 37°C in 5% CO2 for 4 h. A 10% sodium dodecyl sulfate solution was added to dissolve the formazan product (crystal violet) reduced by the cells and reacted for 2 h. The optical density was measured at 570 nm using a microplate reader (Multiskan FC; Thermo Fisher Scientific). For spleen cells, Cell Counting Kit-8 (DOJINDO Laboratories) solution was added for 4 h. The optical density was measured at 450 nm using a microplate reader. To measure cell viability, DCs were cultured and treated in 6-well plates under the same conditions as described above. A trypan blue exclusion assay was performed by counting viable and dead cells, and the viability was calculated.
Measurement of cytokines
DCs were cultured in 96-well plates at a density of 2 × 105 cells/ml and treated with DSF and LPS for 2 days. The supernatants of treated cells were used for enzyme-linked immunosorbent assay (ELISA). ELISA kits for tumor necrosis factor (TNF)-α, IL-12 (Invitrogen), and IL-6 (BioLegend) were used according to the manufacturer's instructions. The optical density was measured at 450 nm using a microplate reader.
Flow cytometry analysis
Flow cytometry analysis was performed to measure apoptosis, surface marker expression, and antigen uptake capability. DCs were treated with DSF and LPS in 6-well plates and incubated for the required days depending on the experimental purpose. DCs were stained with annexin V-fluorescein isothiocyanate (FITC) (Thermo Fisher Scientific) and propidium iodide (PI; Sigma) for the cell apoptosis assay. DCs were treated for 1 day and washed with annexin V-binding buffer. Annexin-FITC and 1.5 µg/ml PI were added and reacted at room temperature for 10 min in the dark. Cells were analyzed using flow cytometry (CytoFLEX LX; Beckman Coulter) and CytExpert software (Beckman Coulter). To measure the expression of activation- or maturation-related surface markers, DCs were treated for 3 days and then stained with allophycocyanin (APC)- or phycoerythrin (PE)-labeled anti-mouse CD11c, FITC-labeled anti-mouse MHC class II (I-Ab), PE-labeled anti-mouse CD40, PerCP Cy5.5-labeled anti-mouse CD54, and APC-labeled anti-mouse CD86 (all from BioLegend) at 4°C for 30 min. TruStain FcX (anti-mouse CD16/32; clone 93; BioLegend) was used to block the Fc receptor. To determine the antigen uptake ability of DCs, after treating DCs for 3 days, FITC-dextran (Sigma-Aldrich) was added and cultured at a concentration of 250 µg/ml at 37°C for 1 h. Incubated cells were stained with APC-labeled anti-mouse CD11c and TruStain FcX. Subsequently, stained cells were analyzed using flow cytometry (LSR Fortessa; BD Biosciences) and the FlowJo software (BD Biosciences).
Mixed lymphocyte reaction (MLR)
To investigate the antigen-presenting capability of the DCs, a MLR was performed. DCs were treated with DSF and LPS for two 2 days prior to co-culture. Before co-culture, DCs were treated with mitomycin C (Sigma-Aldrich) at a concentration of 50 µg/ml for 30 min to prevent spontaneous activation during co-culture. Allogeneic spleen cells were harvested from the BALB/c mice. Treated DCs and allogeneic spleen cells were co-cultured at concentrations of 1 × 105 cells/ml and 2 × 106 cells/ml, respectively, in 96-well plates. After 5 days, the Cell Counting Kit-8 (DOJINDO Laboratories) solution was added for 4 h. The optical density was measured at 450 nm using a microplate reader.
Inhibition of GM-CSF signaling
DCs were harvested and baricitinib (Tocris Bioscience) was pretreated for 4 h at a final concentration of 200 nM. 10 ng/ml GM-CSF was added and DCs were incubated for 24 h.
Quantitative real-time PCR (qPCR)
DCs were cultured in medium only, without cytokines for 48 h, and then treated with GM-CSF for 24 h. After treatment, total RNA was isolated from DCs using an easy-spin Total RNA Extraction Kit (iNtRON Biotechnology). cDNA was synthesized from the RNA samples using the Power cDNA Synthesis Kit (iNtRON Biotechnology) containing oligo (dT) and random hexamer primers. qPCR reactions using AccuPower 2X GreenSta qPCR Master Mix (Bioneer) were run in a Thermal Cycler Dice Real-Time PCR Single (Takara Bio). The primers used for XBP-1 qPCR assay are forward primer 5′-CCTGAGCCCGGAGGAGAA-3′ and reverse primer 5′-CTCGAGCAGTCTGCGCTG-3′ (Bioneer). The relative amounts of mRNAs were calculated from the comparative threshold cycle (Ct) values using β-actin as a control.
Statistical analysis
Statistical analyses were performed using Prism 9 software (GraphPad Software). Data are expressed as mean ± standard deviation. All experiments were performed at least thrice. Statistical significance was analyzed using unpaired t-test, ordinary one-way or two-way ANOVA followed by Dunnett's multiple comparisons test, Šídák's multiple comparisons test, or Tukey's multiple comparisons test. Statistical significance was set at p < 0.05. *, **, ***, **** indicate p < 0.05, 0.01, 0.001, 0.0001 compared to the control, respectively. ##, ###, #### represents the p < 0.01, 0.001, 0.0001 between the different groups at the same DSF concentration. p < 0.05 was considered statistically significant.
Go to :
RESULTS
DSF had less effect on the metabolic activity of DCs than on BMs and spleen cells
To evaluate the effect of DSF on DCs, BMs, and spleen cells, the cells were treated with DSF and LPS (Fig. 1). The metabolic activity of DCs showed no significant changes in the absence or presence of LPS. 1 µg/ml of DOX was used as a positive control and the cellular metabolic activity significantly decreased in 1 µg/ml DOX-treated DCs. Unlike on DCs, 1 µM DSF decreased the metabolic activity of BMs and spleen cells. In the presence of LPS, the metabolic activity of the BMs and spleen cells was significantly decreased in DSF 0.2 µM. In particular, the metabolic activity of spleen cells decreased by 67.7% compared to the control at DSF 0.2 µM in the presence of LPS, while that of BMs decreased by 15.5%. Taken together, DSF had less effect on the metabolic activity of DCs compared to that of BMs and spleen cells.
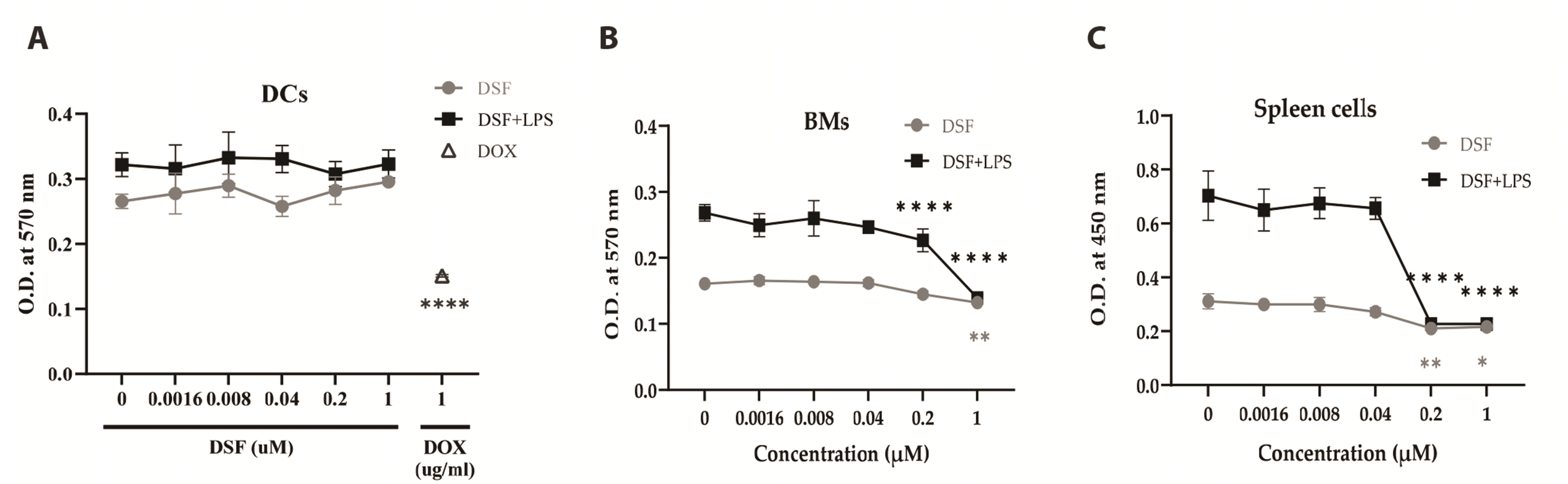 | Fig. 1The effect of DSF on the cellular metabolic activity.(A) DCs. (B) BMs. (C) Spleen cells. Cells were cultured in a 96-well plate with 0 to 1 µM of DSF with or without LPS. Concentrations of LPS were 0.1 µg/ml on DCs and BMs, and 1 µg/ml on spleen cells. 1 µg/ml of DOX was used as a positive control on DCs. The MTT assay was performed at 3 days after the treatment. The optical density (O.D.) was measured at 570 nm by using a microplate reader. Results are presented as mean ± SD, and statistical significance was performed by two-way ANOVA followed by Dunnett’s multiple comparisons test. *, **, **** indicate p < 0.05, 0.01, 0.0001 respectively compared to control cells (DSF 0 µM). DSF, Disulfiram; DCs, dendritic cells; BMs, bone marrow cells; LPS, lipopolysaccharide; DOX, doxorubicin; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenylltetrazolium bromide.
|
DSF has little effect on apoptosis and cell viability of DCs
To analyze cell death, DCs were treated with DSF and LPS and incubated for 1 day. Annexin V-FITC/PI staining was then performed (Fig. 2A, B). Cell death was classified as necrosis (annexin V–/PI+), early apoptosis (annexin V+/PI–), and late apoptosis (annexin V+/PI+). The distribution and ratio of cell death did not significantly change as DSF and LPS were treated on DCs. Trypan blue exclusion assay performed after treating the cells for 3 days significantly decreased the number of 0.2 µM (Fig. 2C). The ratio of dead to live cells increased as the total number of cells decreased to 0.2 µM. It increased by 26.6% and 9.7% in the presence and absence of LPS at 0.2 µM DSF compared to the control group. However, the total cell viability did not change significantly (Fig. 2D).
 | Fig. 2DSF has little effect on apoptosis and cell viability.After 24 h of treatment, the DCs were stained with Annexin V-FITC and PI. (A) The quadrants of dot plot indicate live cells (annexin V–/PI–), the cells in early apoptosis (annexin V+/PI–), late apoptosis (annexin V+/PI+), and necrosis (annexin V–/PI+). (B) Statistical analysis was performed based on the results of the four experiments conducted under the same conditions. A trypan blue exclusion assay was performed after 3 days of treatment. (C, D) As a result of four experiments under the same conditions, the net number of live and dead cells was counted. The viability was calculated by the ratio of live cells to total cells. Results are presented as mean ± SD, and statistical significance was performed by two-way ANOVA followed by Tukey’s multiple comparisons test (B, C) or Dunnett’s multiple comparisons test (D). * indicates p < 0.05 compared to control DCs (DSF 0 µM). DSF, Disulfiram; DCs, dendritic cells; LPS, lipopolysaccharide; FITC, fluorescein isothiocyanate; PI, propidium iodide.
|
DSF has little effect on the maturation of DCs
DCs were treated with DSF and LPS for 3 days and incubated with FITC-dextran to determine how the antigen uptake capability changed (Fig. 3A, B). In addition, the expression levels of surface markers on DCs were measured to determine whether DSF affected on DC activation and maturation (Fig. 3C). Antigen uptake capacity was strongly reduced only in the presence of LPS, and no significant decrease was observed as the concentration of DSF increased. Similarly, the expression of surface markers on DCs was significantly increased only in the presence of LPS, and there was no significant change induced by DSF. In summary, DSF did not significantly affect the maturation of LPS-treated DCs.
 | Fig. 3DSF does not affect the maturation of LPS-treated DCs.To evaluate the effect of DSF on the maturation of DCs, antigen-uptake capability and expression of maturation-related surface markers of DCs were analyzed. After 3 days of treatments, the DCs were incubated with FITC-dextran for 1 h or stained with the marker-specific antibodies for 30 min as described in the “Methods” section. Anti-CD11c antibody was used for gating DCs. (A) Numbers of histograms represent the geometric mean of fluorescence intensity. (B, C) For statistical analysis of three independent experiments, the mean fluorescence intensity detected in the control DCs was set to 100%. Results are presented as mean ± SD, and statistical significance was performed by two-way ANOVA followed by Šídák’s multiple comparisons test. * indicates p < 0.05 compared to control DCs (DSF 0 µM). DSF, Disulfiram; DCs, dendritic cells; LPS, lipopolysaccharide; FITC, fluorescein isothiocyanate.
|
DSF lowers the production of IL-12/23 (p40)
IL-12/23 (p40), IL-6, and TNF-α, representative cytokines secreted from DCs, were analyzed using ELISA (Fig. 4). As a result, only IL-12/23 (p40) production markedly decreased at 0.2 and 1 µM. DSF also reduced the production of IL-6 from 0.2 µM, but not significantly. This result suggests that the production of IL-12/23 (p40) in LPS-treated DCs is sensitive to DSF, compared to that of IL-6 and TNF-α.
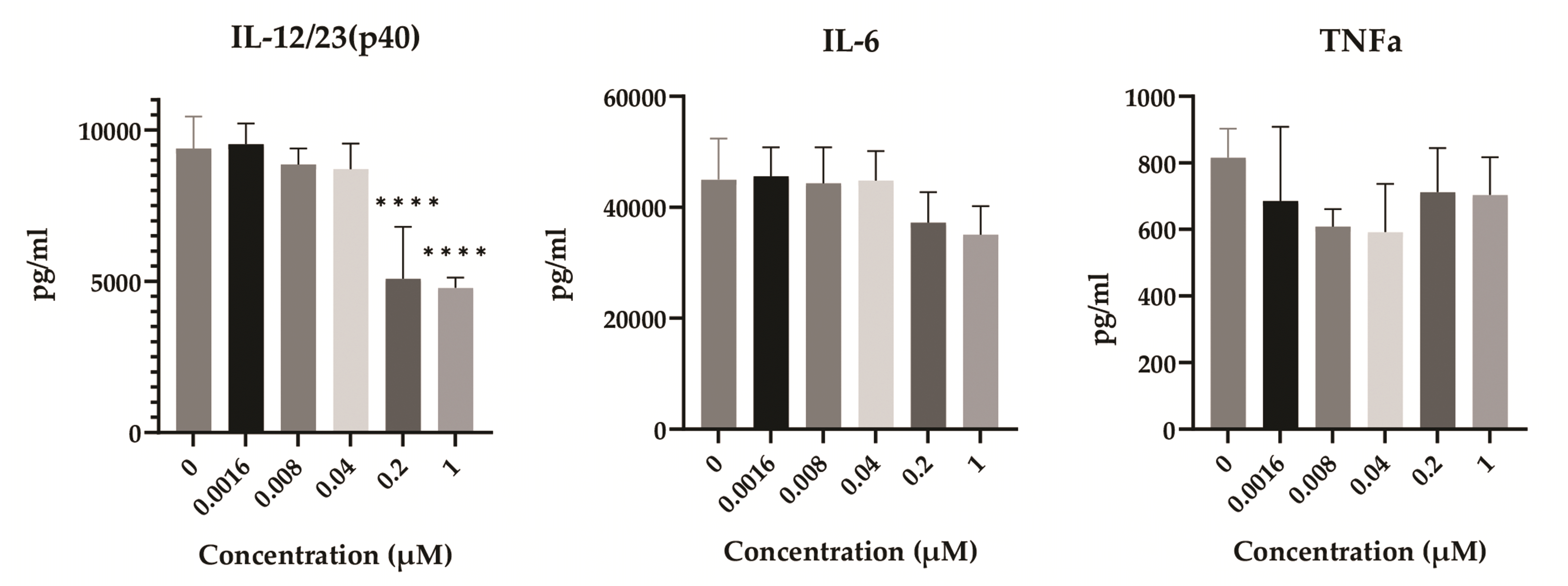 | Fig. 4The effect of DSF on the production of immune-related cytokines.After 2 days of treatments, the supernatant of treated DCs was collected and used for ELISA. The optical density was measured at 450 nm by using a microplate reader. Results are presented as mean ± SD, and statistical significance was performed by ordinary one-way ANOVA followed by Dunnett’s multiple comparisons test. **** indicates p < 0.0001 compared to control DCs (DSF 0 µM). DSF, Disulfiram; DCs, dendritic cells; IL, interleukin; TNF, tumor necrosis factor.
|
DSF does not alter the antigen-presenting capability of DCs
To investigate the effects of DSF on the antigen-presenting capability of DCs, the treated DCs, and allogeneic spleen cells were co-cultured (Fig. 5). As DCs matured by LPS, the antigen presentation ability increased in the LPS treatment group, but there was no marked alteration depending on the concentration of DSF. This result demonstrated that DSF does not affect the antigen-presenting ability of DCs.
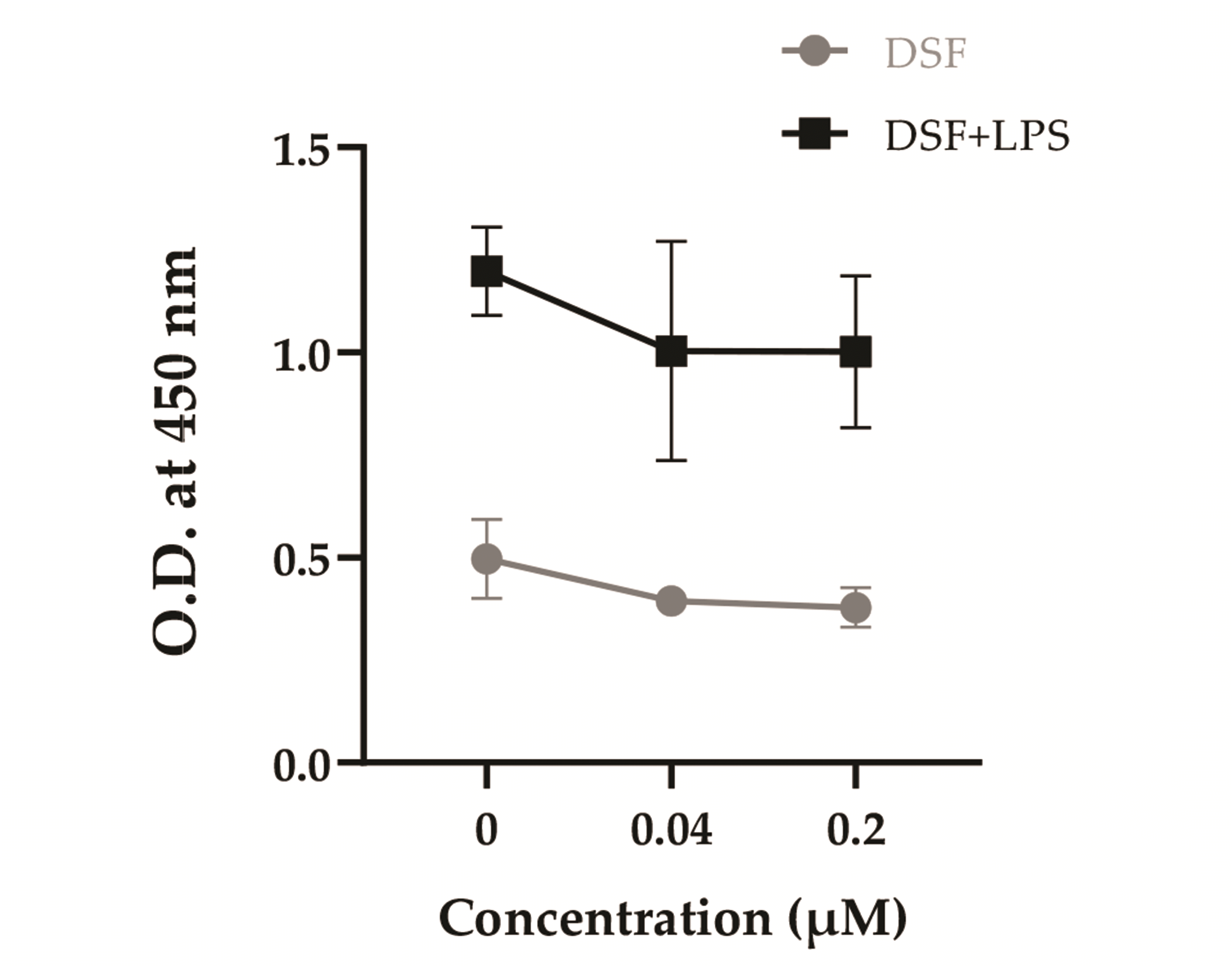 | Fig. 5Effect of DSF on the antigen-presenting capability of DCs.After 3 days of DSF and LPS treatment, the DCs were incubated with 50 µg/ml mitomycin C for 30 min and co-cultured with allogeneic spleen cells. After 5 days, CCK-8 assay was performed and the optical density (O.D.) was measured at 450 nm by using a microplate reader. Results are presented as mean ± SD, and statistical significance was performed by two-way ANOVA followed by Dunnett’s multiple comparisons test. DSF, Disulfiram; DCs, dendritic cells; LPS, lipopolysaccharide.
|
GM-CSF is closely connected to the DCs' resistance to DSF
We further investigated what makes the resistance of DCs to DSF (Fig. 6A). DCs were divided into three groups; GM-CSF treated, GM-CSF not treated and GM-CSF signaling blocked. DCs were cultured without GM-CSF for 24 h (Baricitinib–/GM-CSF–) or pretreated with baricitinib before given GM-CSF (Baricitinib+/GM-CSF+). In both groups, the metabolic activity of DCs decreased in a DSF concentration-manner (Fig. 6B). At 1 µM DSF, the metabolic activity decreased to 85.9% in Baricitinib–/GM-CSF– DCs and 67.8% in Baricitinib+/GM-CSF+ DCs compared to the control group (0 µM DSF), whereas no significant decrease in Baricitinib–/GM-CSF+ DCs. Moreover, when compared to the Baricitinib–/GM-CSF+ DCs, the metabolic activity was significantly decreased from 0.2 µM DSF in Baricitinib–/GM-CSF– and Baricitinib+/GM-CSF+ DCs. Next, RT-qPCR demonstrated that GM-CSF increased the expression of XBP-1 (Fig. 6C). IL-4 also increased the expression of XBP-1. However, when DCs were treated with GM-CSF and IL-4, the increase of XBP-1 expression was similar to that of GM-CSF alone (data not shown).
 | Fig. 6GM-CSF is responsible for the resistance of DCs to DSF.(A) Schematic overview of the experiment. (B) DCs were pretreated with or without baricitinib for 4 h. 10 ng/ml of GM-CSF was added (or not) and DCs were incubated for 24 h. 0–1 µM of DSF were treated and incubated for 48 h. The metabolic activity was measured by MTT assay. (C) DCs were cultured in a culture medium without GM-CSF + IL-4 for 48 h. The expression of XBP-1 in DCs was measured using qPCR with or without GM-CSF treatment for 24 h. Results are presented as mean ± SD. Statistical significance was performed by two-way ANOVA with Šídák’s multiple comparisons test and Dunnett’s multiple comparisons test (B), or unpaired t-test (C). **, **** indicate p < 0.01, 0.0001 respectively, compared to control DCs (DSF 0 µM). #, ##, #### indicate p < 0.1, 0.01, 0.0001 compared between the groups at the same concentration of DSF. GM-CSF, granulocyte-macrophage colony-stimulating factor; DSF, Disulfiram; DCs, dendritic cells; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenylltetrazolium bromide; qPCR, quantitative real-time PCR; IL, interleukin.
|
Go to :
DISCUSSION
DSF is used as an inhibitor of aldehyde dehydrogenase in alcoholism [1]. Recently, DSF has been considered a candidate anticancer agent [2]. The anticancer research regarding DSF is being conducted more actively as the synergy between DSF and Cu2+ has been revealed [8]. In this study, we evaluated the effect of DSF on the survival and function of DCs, which are the most competent antigen-presenting cells in immunity. Cancer therapies, including chemotherapy, radiation therapy, and immune checkpoint therapy can affect or even depend on DCs [13]. Proper knowledge of a drug’s effects on immune cells is essential for evaluating anticipated side effects and whether it can be used in combination with other drugs. To confirm the direct effects of DSF on DCs, we focused on in vitro study using cultured DCs. Since DCs have various subtypes by tissues and DSF can affect on diverse cells when treated in vivo, it is difficult to investigate the direct effect of DSF on DCs in vivo.
Based on the MTT assay, DSF decreased the metabolic activity of BMs and spleen cells but not that of DCs (Fig. 1). The half maximal inhibitory concentration (IC50) of DSF against DCs was 2.24 µM, which is much higher than that of BMs and spleen cells (0.15 µM and 0.48 µM, respectively). Moreover, the IC50 of DSF in DCs was higher than that in the cancer cell lines. In MCF7 and Raji cell lines, IC50 of DSF was 0.5 µM and 0.79 µM each [7,14]. When adding cooper, IC50 of DSF decreased in cancer cells; 0.085 µM in Raji cell lines [7] and 0.2–0.5 µM in breast cancer cell lines [6]. To measure cell death in the stages from early apoptosis to necrosis, we stained the annexin V/PI 1 day after treatment of DCs with DSF and LPS (Fig. 2A, B). Cell viability analysis was performed 3 days after the treatment, under the same conditions as most other functional analyses (Fig. 2C, D). Apoptosis was slightly increased by DSF 0.2 µM. The trypan blue exclusion assay revealed that the total number of DCs was decreased by DSF, but the viability was not. It is suggested that the damage of DSF on DCs may affect survival and the total cell number at the end point, but not cellular metabolic activity; that is, DCs are resistant to DSF and have lower cellular toxicity than BMs and spleen cells.
When DCs capture antigens, they undergo maturation. During maturation, they lose their antigen uptake ability but acquire costimulatory functions and eventually induce an antigen-specific primary T-cell response [9]. We investigated how DSF alters this sequence of processes in DCs. MHC class II molecules are essential antigen-presenting molecules that present antigens to T cells. CD86 acts as a co-stimulator of T cell activation, CD40 is related to T cell-mediated activation, and CD54 is related to cell-cell adhesion [15]. DSF did not affect the expression of these surface markers or the antigen uptake capability (Fig. 3). In addition, DSF did not decrease antigen-presenting capability of DCs in the co-culture with allogeneic spleen cells (Fig. 5). DSF can exert its medicinal properties without impairing DC function.
DCs secrete cytokines such as IL-6, TNF-α, and IL-12. IL-6 and TNF-α are proinflammatory cytokines [16]. IL-12, a representative cell-mediated immunity cytokine of DCs, has been overexpressed in many autoimmune diseases. IL-23 has also been considered a key cytokine in autoimmunity [17,18]. Because of the rapid decrease in the production of IL-12/23 (p40) (Fig. 4), rather than other cytokines, DSF is expected to be positively utilized in the treatment of several diseases targeting IL-12/23 (p40). However, since we measured p40, a common subunit of IL-12 and IL-23, we need to determine which of the two cytokines had a greater effect on the results. Moreover, no significant decrease in IL-6 and TNF-α production above 0.2 µM DSF indicates that the rapid decrease in IL-12/23 (p40) was not due to cell death.
What makes DCs relatively resistant to DSF, unlike BMs and spleen cells? Immune cells undergo apoptosis during their development, and this is essential for maintaining immune homeostasis [19]. Cytokines are among the important regulators of immune cell development and affect cell death [20,21]. Apoptosis of DCs is thought to be controlled by a balance between Bcl-2-related proteins, rather than through the action of death receptor proteins such as Fas [22]. On the other hand, Fas and TNFR are markedly expressed in T cells and B cells. Different immune cell types have different regulators that affect differentiation and cell death, which may explain their differential resistance to DSF.
Although there may be a variety of factors that influence the survival of DCs, we focused the main growth factor of DCs first. GM-CSF is a well-known positive regulator of DC development [23] which helps DCs homeostasis by inhibiting Bim-dependent apoptosis [24]. GM-CSF signaling is primarily regulated by Janus kinase 2 (JAK2). Baricitinib, a selective JAK1/2 inhibitor [25], was used to investigate whether GM-CSF affects the resistance of DCs on DSF. There was a significant difference in the resistance of DCs to DSF depending on the presence of GM-CSF for 24 h before DSF treatment (Fig. 6B). Considering that all DCs were exposed to GM-CSF during the development, these data show how GM-CSF significantly effects on DCs. Among the many subfactors stimulated by GM-CSF, we further examined XBP-1. XBP-1 is a transcription factor essential for DC growth and survival. Specifically, XBP-1 alleviates the unfolded protein response by maintaining endoplasmic reticulum (ER) homeostasis and preventing cell death due to ER stress [26]. To minimize the effect of GM-CSF exposed on DCs during the culture, DCs were starved for 2 days without any cytokines in the growth medium before the experiment. GM-CSF significantly increased the expression of XBP-1 in DCs. Taken together, these data verified that GM-CSF upregulates XBP-1 and plays a role in the resistance of DCs to DSF.
In conclusion, DSF is less cytotoxic to DCs than to BMs and spleen cells and has little effect on the function of DCs. Most anticancer drugs have immunosuppressive side effects, especially if they are toxic to immune cells. As DSF has an anticancer effect but is less toxic to DCs, it may alleviate the side effects of immunosuppression by reducing the dose of anticancer drugs when used in combination with other anticancer drugs. Given that DSF reduces IL-12/23 (p40) production of DCs, it can be a treatment option for autoimmune inflammatory diseases. This study provides experimental evidences that DSF can be used as a new repurposing drug for cancer therapy and immune diseases, particularly when DC damage needs to be reduced.
Go to :
 XML Download
XML Download