

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
Functional high voltage-gated calcium channels (HGCCs) such as L-type, P/Q-type, N-type, and R-type channels, comprise a pore-forming α subunit and ancillary β, α2δ, and γ subunits [1,2]. The β subunit acts in channel gating and activation-inactivation kinetics, besides trafficking channel proteins to the plasma membrane in many cell types [3]. In cardiomyocytes, the β subunit, participating in functional L-type HGCCs, is required for β-adrenergic receptor-mediated modulation [4]. However, since the structure of the protein-protein interaction between the pore-forming α subunit and the ancillary β subunit was discovered [5-7], the disturbance of the α-β interaction has been considered a target to regulate HGCC-mediated normal or abnormal functions. For example, an 18-amino-acid peptide derived from the α-interaction domain (AID) in the cytoplasmic domain I-II linker of the α subunit can disrupt the α-β subunit interaction and affect the function of HGCCs [8-12].
We previously showed that two different peptides derived from the AID-binding sequences in the guanylate kinase domain of the β subunit significantly decreased the peak amplitudes of voltage-pulse-activated Ca2+ currents in trigeminal ganglion (TG) neurons [13]. Moreover, if the peptides are attached to the membrane-permeable HIV transactivator of transcription peptide, they reduce blood pressure and peak amplitudes of excitatory postsynaptic currents upon the intravenous application of an external bath in rats [13]. This suggests that peptides disturbing the α-β subunit interaction inhibit or modulate the physiological functions of intact HGCCs.
While the two peptides previously tested are derived from the α3 helix and β10 sheet-α9 helix parts of the AID-binding sequence in the β subunit [13], we further tested whether peptide fragments derived from the β9 sheet-α6 helix, another important region of the α-β interaction, affect function of HGCCs in the TG neurons and arterial blood pressure of rats.
Go to : 
METHODS
Origins and synthesis of peptides
The peptide fragments used in this study were derived from voltage-gated calcium channel auxiliary subunits β1, β2, β3, and β4; their protein accession numbers are P54283.1, Q8VGC3.2, P54287.1, and Q8R0S4.2, respectively (Table 1). The peptide sequences are listed in Table 1, and most of them are patented in Korea (Patent no. 10-2425548, year 2022). All the peptides were synthesized by a peptide synthesis company (Peptron, Inc.), and the purity of each peptide (> 95%) was determined by high-performance liquid chromatography (according to the information provided by the company).
Table 1
Sequences of 16-mer peptides derived from voltage-gated calcium channel β subunits
![]()
Whole-cell recordings of Ca2+ currents from TG neurons
All procedures with animals were approved by the Institutional Animal and Use Committee of Kyungpook National University (Approval no. 2019-008-2). TG neurons were collected from the TG of Sprague-Dawley rats (7–16 day-old; male or female). Initially, the ganglia were incubated at 37°C for 15 min in prewarmed L-15 media containing collagenase D (1 mg/ml; Roche Diagnostics GmBH). After trituration, ganglion neurons were harvested by washing twice with 1× phosphate-buffered saline and plated on poly-D-lysine/laminin-coated coverslips (diameter, 25 mm) in DMEM (Sigma) containing 10% fetal bovine serum and 1% penicillin/streptomycin. Neurons were used for whole-cell recordings within 1–6 h after maintaining at 37°C with 5% CO2.
To record Ca2+ currents in the isolated TG neuron, whole-cell voltage-clamp recordings were performed with 3–5 MΩ heat-polished borosilicate patch pipettes (TW150F; WPI) on the inverted microscope (IX51; Olympus) at room temperature (24°C–26°C). The internal solution was composed of CsCl (135 mM), HEPES (10 mM), EGTA (1 mM), EDTA (1 mM), and MgCl2 (4 mM), and was adjusted to pH 7.2 with 20% tetraethylammonium (TEA) hydroxide. The bath solution contained TEA-Cl (135 mM), CaCl2 (2 mM), HEPES (10 mM), and tetrodotoxin (TTX) (0.0001 mM) (pH 7.2; adjusted by with 20% TEA hydroxide). TEA and TTX was added to the internal and/or external solution to suppress the induction of K+ or Na+ currents, respectively. The TG neurons were held at holding potential (Vh) of –60 mV, and voltage-activated currents were evoked every 15 sec by depolarizing voltage steps (from –50 mV to +40 mV in 10 mV increment for 250 or 500 ms). The recorded currents were amplified with Axopatch 1D (Molecular Devices) and filtered at 2 kHz (sampling rate, 5 kHz) using pClamp software (Molecular Devices). Transients due to whole-cell capacitance and series resistance were cancelled, and leak currents were subtracted using scaled hyperpolarizing currents (-P/4 protocol). The peak amplitudes of currents were adjusted by the whole-cell capacitance (pA/pF), and neurons with > 10 pA/pF were included for analysis. The peptides were applied through the internal solution.
Measurement of arterial blood pressure in anesthetized rats
Arterial blood pressure and peptide solution infusion were measured using a catheter system in the femoral artery and femoral vein, respectively, in Sprague-Dawley rats (330–430 g) of both sexes. Initially, the rats were anesthetized with isoflurane (2%) vaporized in oxygen (1%–1.5%), and the anesthesia was maintained by urethane (1 g/kg, intraperitoneal injection) and isoflurane (0.8%–1%) via snout mask. To expose the femoral neurovascular bundle, the skin in the femoral region was incised, and the soft tissue was dissected. The femoral sheath was opened using blunt dissection directed along its long axis between the femoral artery and vein (dark red). The femoral artery was retracted laterally, and the femoral vein was retracted medially. Then, two pieces of silk (4-0) were inserted under the artery just below the arteria profunda femoris, and one piece after a loose ligature (for upcoming proximal ligation) was pulled toward the body and the other was tightly ligated by a triple knot as far as possible toward the leg (distal ligation). Similarly, two pieces of silk (4-0) were inserted under the vein below the vena profunda femoris, and a loose ligature for the upcoming proximal ligation and a distal tight ligature were made. To insert catheters (PE50 tube) into the femoral artery and femoral vein, a small incision using micro-dissecting scissors was made approximately 2 mm proximal to the distal ligation. The catheter, pre-filled with a saline solution containing 300 U/L of heparin with a syringe (1 ml), was then partly inserted through the incision. The pulled proximal ligature was then loosened, and the catheter was fully inserted (> 1 cm). Then, proximal ligation was accomplished using a triple knot around the artery and catheter or the vein and catheter to secure the catheter to the vessel. Catheter patency was further confirmed by identifying the blood inside the inserted catheter and flushing the heparinized saline solution.
The catheter inserted through the femoral artery was connected to a pressure transducer placed at the same height as the heart of the rat. The transducer was further connected to a PowerLab data acquisition system (ADInstruments). Arterial blood pressure data were recorded and analyzed using LabChart (version 7; ADInstruments). From the cyclic records of arterial blood pressure, systolic pressure was identified by the peak and diastolic pressure by the valley between peaks. Mean arterial pressure was calculated by adding one-third of the difference between the systolic and diastolic pressures to the diastolic pressure. Heart rate was determined as the number of cycles per minute. The ∆ blood pressure was calculated by subtracting the blood pressure after the injection of a peptide solution from the blood pressure before the injection.
Non-invasive measurement of arterial systolic blood pressure in non-anesthetized rats
Rats were habituated to the apparatus and environment to measure their systolic pressure. A rat was warmed at 30°C for 30 min in an isothermal container and constrained by an acryl holder with a tail-cuff sensor. Systolic pressure was measured in the non-anesthetized state using a non-invasive tail-cuff blood pressure system (Model 47; IITC Life Science). The average of three consecutive measurements was calculated. Peptide solutions or saline was administered through the tail vein of rats after light isoflurane anesthesia in a closed container.
Intrathecal catheter insertion and formalin test
A PE-10 catheter was inserted into the L3-4 lumbar subarachnoid space of a 7–8 week-old rat (male or female) under deep isoflurane anesthesia (2.5%–3%). The rat was recovered for a week, and was subjected to formalin test after adapting to the test environment for 0.5–1 h. The peptide solution (10 µl) was intrathecally administered through the catheter 30 min before the formalin test. The formalin test was conducted using the formalin solution (5%, 50 µl) that was intradermally injected into the plantar surface in one of rat hindpaws (typically left hindpaw). Immediately after the formalin injection, pain behaviors, such as paw withdrawal or licking, were counted and presented in 5-min blocks up to 65 min.
Statistical analysis
Statistical comparisons were performed using an unpaired Student’s t-test (*p < 0.05, **p < 0.01). Data are reported as mean ± standard error of the mean (SEM).
Go to :
RESULTS
Peptides derived from HGCC β subunits decreased arterial blood pressures
In this study, we tested the 16-mer peptide fragments derived from HGCC β1 (cacb1), β2 (cacb2), β3 (cacb3), and β4 (cacb4) subunits to determine whether they affected the function of HGCCs and, therefore, arterial blood pressure in rats. In an electrophysiological experiment using whole-cell voltage-clamp recordings in acutely isolated TG neurons [13], the synthesized peptides (water solubility, > 10 mg/ml) did not change the amplitudes of voltage-activated Ca2+ current upon their intracellular application through recording patch pipettes (Fig. 1). However, when the peptides were intravenously administered at 0.15 mg/kg (volume, 0.2 ml), the arterial blood pressure from the femoral artery started to decrease and peaked within a minute (the case of β1 subunit, cacb1(344–359); Fig. 2A). As the dose was increased to 1 mg/kg, the magnitude of the reduction in arterial pressure became more intense and longer in duration (Fig. 2A), and pulse pressure slightly decreased (Fig. 2B). Thus, the effect of cacb1(344–359) on systolic pressure was dose-dependent (Fig. 3A), and the systolic, diastolic, and mean arterial pressure were decreased by 23.7 ± 10.4 mmHg, 22.0 ± 8.6 mmHg, and 22.6 ± 9.2 mmHg, respectively, at a dose of 0.5 mg/kg in three rats, and the heart rate was slowed by 36.0 ± 22.4 beats per minute (Fig. 3B). However, the arterial blood pressure remained unchanged by intravenous injection of either saline or another 16-mer control peptide [cacb1(389–404)], derived from a different part of the β1 subunit (Fig. 2C) (average changes from five rats at 0.5 mg/kg: systolic pressure, −0.4 ± 1.2 mmHg; diastolic pressure, −0.2 ± 1.2 mmHg; mean arterial pressure, −0.3 ± 1.1 mmHg; Fig. 3B). Peptides from the β2 [cacb2(392–407)], β3 [cacb3(292–307)], and β4 [cacb4(333–348)] subtypes, which are the same part but have slightly different amino acid sequences compared to β1, also decreased arterial blood pressure in rats (Fig. 3A). The effects of these 16-mer peptides on systolic blood pressure were dose-dependent; the effects on systolic, diastolic, and mean arterial pressure were significantly different at 0.5 mg/kg compared to that of the control cacb1(389–404) peptide (Fig. 3B).
 | Fig. 1Peptides derived from high voltage-gated calcium channel β subunits do not change the peak amplitude of high voltage-activated Ca2+ currents recorded in the acutely isolated trigeminal ganglion neurons.(A) Voltage step (from –60 mV to –50–40 mV, 500 ms)-activated Ca2+ currents were recorded in the isolated trigeminal ganglion (TG) neurons with the internal whole-cell patch solution containing β1 [cacb1(344-359)], β2 [Cacb2(392-407)], β3 [cacb3(292-307)], and β4 [cacb4(333-348)] subunits-derived peptides. (B) Summarized current-voltage relationships depicted by whole-cell recordings with internally applied individual or all peptides. Numbers indicate the number of recorded TG neurons. Values are presented as mean ± SEM.
|
 | Fig. 2Decrease of the arterial blood pressure (BP) by a peptide fragment derived from high voltage-gated calcium channel (HGCC) β1 subunit in anesthetized rats.(A) Intravenous injection (0.15 mg/kg, 0.2 ml, IV) of a 16-mer peptide IKITSPKVLQRLIKSR, which is a part of HGCC β1 subunit [cacb1(344–359)], decreased the arterial BP from the femoral artery. The intravenous injection was performed through a catheter inserted into the femoral vein. The decreasing effect of cacb1(344–359) on arterial BP was more intense and longer when the amount of peptide was increased to 1 mg/kg. (B) The decreased systolic, diastolic, mean arterial and pulse pressure are shown in the extended version of the recording. Numbers indicate the time sampled on the pressure recording shown in A. (C) Intravenous injection of saline or the 16-mer peptide LDENQLEDACEHLAEY, which is a different part of the β1 subunit [cacb1(389–404)] and was used as a control, did not change arterial BP.
|
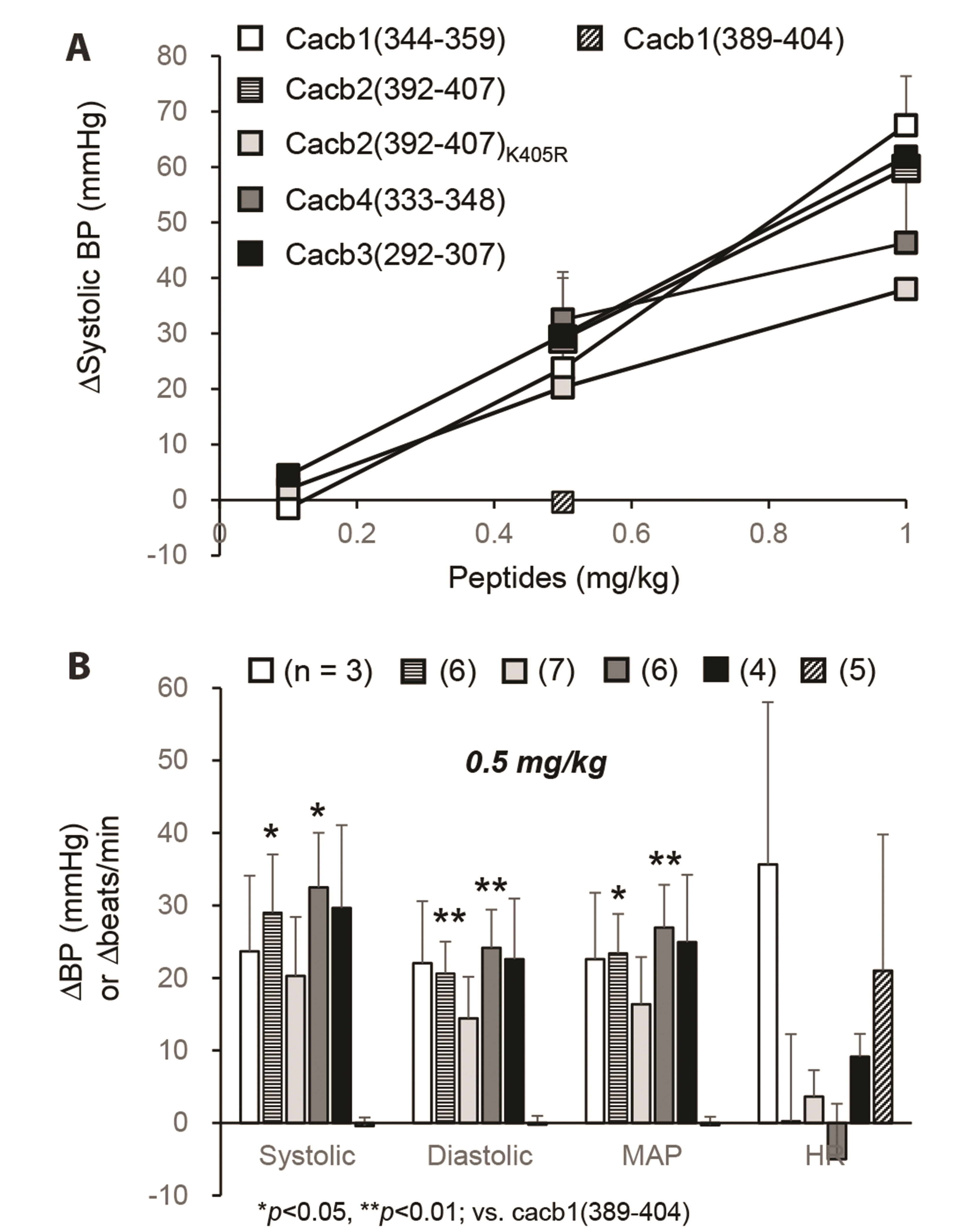 | Fig. 3Decrease of the arterial blood pressure (BP) by peptide fragments of high voltage-gated calcium channel (HGCC) β2, β3, and β4 subunits, which are similar to β1.(A) Summary of the decreases of systolic BP by various 16-mer peptides originated from HGCC β1 [cacb1(344–359)], β2 [cacb2(392–407)], β3 [cacb3(292–307)], and β4 [cacb4(333–348)] subunits. The effects of the 16-mer peptides were dose-dependent, except for that of the control peptide cacb1(389–404). (B) Average decreases in systolic, diastolic, and mean arterial pressure (MAP), and heart rate (HR). Numbers in parentheses indicate the number of rats used. *p < 0.05 and **p < 0.01, vs. cacb1(389–404).
|
Amino acid sequence difference did not affect blood pressure-reducing effects
The peptide sequence comparison showed that cacb2(392–407) had mutations at I344 (to V) and T347 (to S) of cacb1(344–359), cacb4(333–348) at I344 (to V), I346 (to V), and T347 (to S) of cacb1(344–359), and cacb3(292–307) at I344 (to V), I346 (to V), T347 (to S), and K357 (to R) of cacb1(344–359) (Table 1). Thus, we tested cacb1(344–359)I344V with a single mutation (to V) only at the I344 position of cacb1(344–359) and cacb1(344–359)I344V/K357R with double mutations at both the I344 and K357 positions, which also decreased arterial blood pressure (Fig. 4). In addition, a peptide with only the K/R mutation at K357 of cacb1(344–359) [IKITSPKVLQRLIRSR, cacb1(344–359)K357R] also decreased arterial blood pressure (Fig. 4). On the other hand, a single K405R mutant of cacb2(392–407) [VKISSPKVLQRLIRSR, cacb2(392–407)K405R; Fig. 3A] decreased arterial blood pressure, similar to the cacb3(292–307) peptide, which is also a K/R mutation from cacb4(333–348). The dose-dependent effects of peptides mutated at the first, fourteenth, or both positions of cacb1(344–359) on systolic pressure are summarized (Fig. 4A). The magnitudes of these peptides mutated from cacb1(344–359) at 0.5 mg/kg were individually plotted in systolic pressure (Fig. 4B), indicating that the reduction in blood pressure was independent of the blood pressure before peptide administration. Additionally, the effects of cacb1(344–359) and its mutant peptides on systolic, diastolic, and mean arterial blood pressure were significantly different from those of other 16-mer control cacb1(389–404) peptide (Fig. 4C). The cacb1(344–359)K357R peptide was selected for further study because it was slightly more potent and consistent at the dose of 0.5 mg/kg than the other peptides and produced relatively constant blood-reducing effect (Figs. 3 and 4).
 | Fig. 4Decrease of the arterial blood pressure (BP) by mutant peptides of cacb1(344–359).(A) Decrease of systolic BP by mutant peptides of cacb1(344–359). The effects of the mutant peptides were dose-dependent, except for that of the control peptide cacb1(389–404). (B) Scatterplot of the systolic pressure before and after intravenous injection of three mutant peptides (I334V, I344V/K357R, and K357R) at the dose of 0.5 mg/kg. All plots are below the dotted line, indicating that the systolic BP decreased after the injection of peptides. The chart legends of (A) and (B) are the same. (C) Histogram of the average pressure decreases by cacb1(344–359) mutant peptides in systolic, diastolic, and mean arterial pressure (MAP), and of the variation in heart rate (HR). Numbers in parentheses indicate the number of rats used. *p < 0.05 and **p < 0.01, vs. cacb1(389–404).
|
N-terminal or C-terminal truncation abolished blood pressure-reducing effect
To further explore the essential amino acids involved in the blood pressure-reducing effects of the peptides, we performed a truncation study in which two amino acids were continuously cut starting from the N-terminal or C-terminal ends of the cacb1(344–359)K357R peptide. Interestingly, the first truncation of two amino acids at either the N-terminal or C-terminal end almost abolished the blood pressure-reducing effect of cacb1(344–359)K357R (Fig. 5); likewise, the second and third truncations by each of the two amino acids negated the effect of peptides. This indicates that all 16 amino acids are necessary for the effect of cacb1(344–359)K357R on blood pressure.
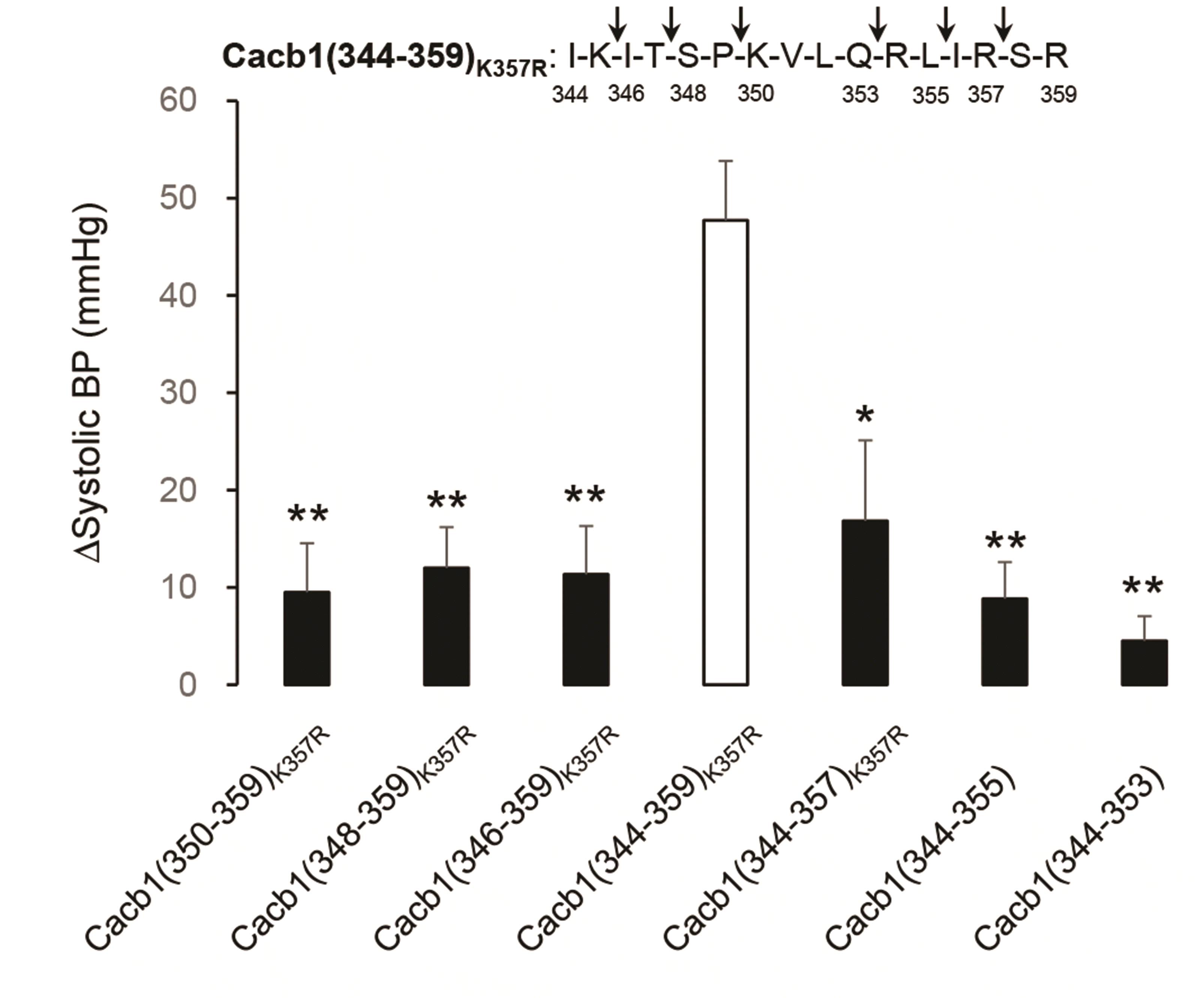 | Fig. 5Loss of the blood pressure (BP)-reducing effects of cacb1 (344–359)K357R by N-terminal or C-terminal truncation.The decrease of BP by cacb1(344–359)K357R was significantly reduced when two amino acids were removed from its N-terminal or C-terminal end. The arrows on the inset above the histogram indicate the cut sites. *p < 0.05 and **p < 0.01, vs. cacb1(344–359)K357R.
|
Palmitic acid conjugation at lysine residues prolonged blood pressure-reducing effect
Despite the effectiveness of cacb1(344–359) and its derivatives in reducing arterial blood pressure, the duration of the effect was relatively short, reaching only 7–8 min (Fig. 6A). Thus, we conjugated palmitic acid, an albumin ligand, to lysine (K) at the 2nd or 7th amino acid position (from the N-terminal) of cacb1(344–359)K357R, respectively producing IK(palmitoyl)ITSPKVLQRLIRSR (named K2-palm) and IKITSPK(palmitoyl)VLQRLIRSR (named K7-palm). Intravenous injection of K2-palm at a dose of 0.5–2 mg/kg substantially prolonged the effect on arterial blood pressure, reaching a maximum duration of over 4 h (Fig. 6B and Fig. 7). K7-palm prolonged the effect at a dose of 2 mg/kg, demonstrating a lower potency than the K2-palm peptide (Fig. 7). In addition, we constructed a fusion peptide of cacb1(344–359)K357R by adding EYEKEYE peptide at its C-terminal end. Then, palmitic acid was conjugated to the lysine side chain of the EYEKEYE sequence (named K20-palm, because of the 20th residue). This peptide also showed a long blood pressure-reducing effect (Fig. 8), but only at a dose of 2 mg/kg, which is four times higher than that of cacb1(344–359)K357R. Therefore, we conclude that the K2-palm peptide is effective in terms of potency and prolongation of the blood-reducing effect.
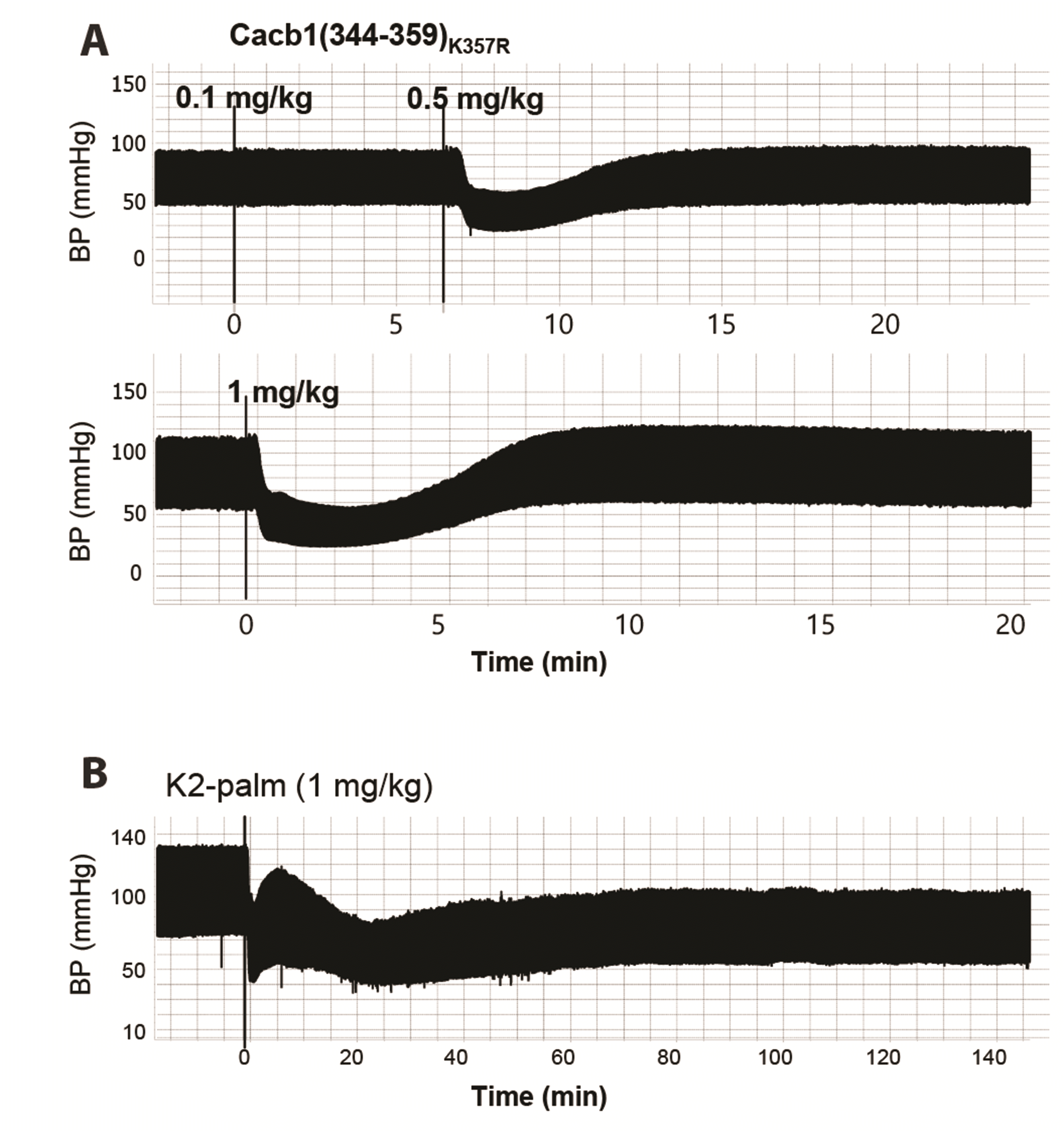 | Fig. 6Prolongation of the blood pressure (BP)-reducing effect by conjugation of palmitic acid with the 2nd lysine of cacb1(344–359)K357R.(A) The mutant peptide cacb1(344–359)K357R caused a dose-dependent decrease of arterial BP with a short duration (~5–8 min). (B) Intravenous injection of cacb1(344–359)K357R conjugated with palmitic acid at its 2nd lysine (K2-palm, 1 mg/kg) prolonged the decrease of arterial BP for more than 2 h.
|
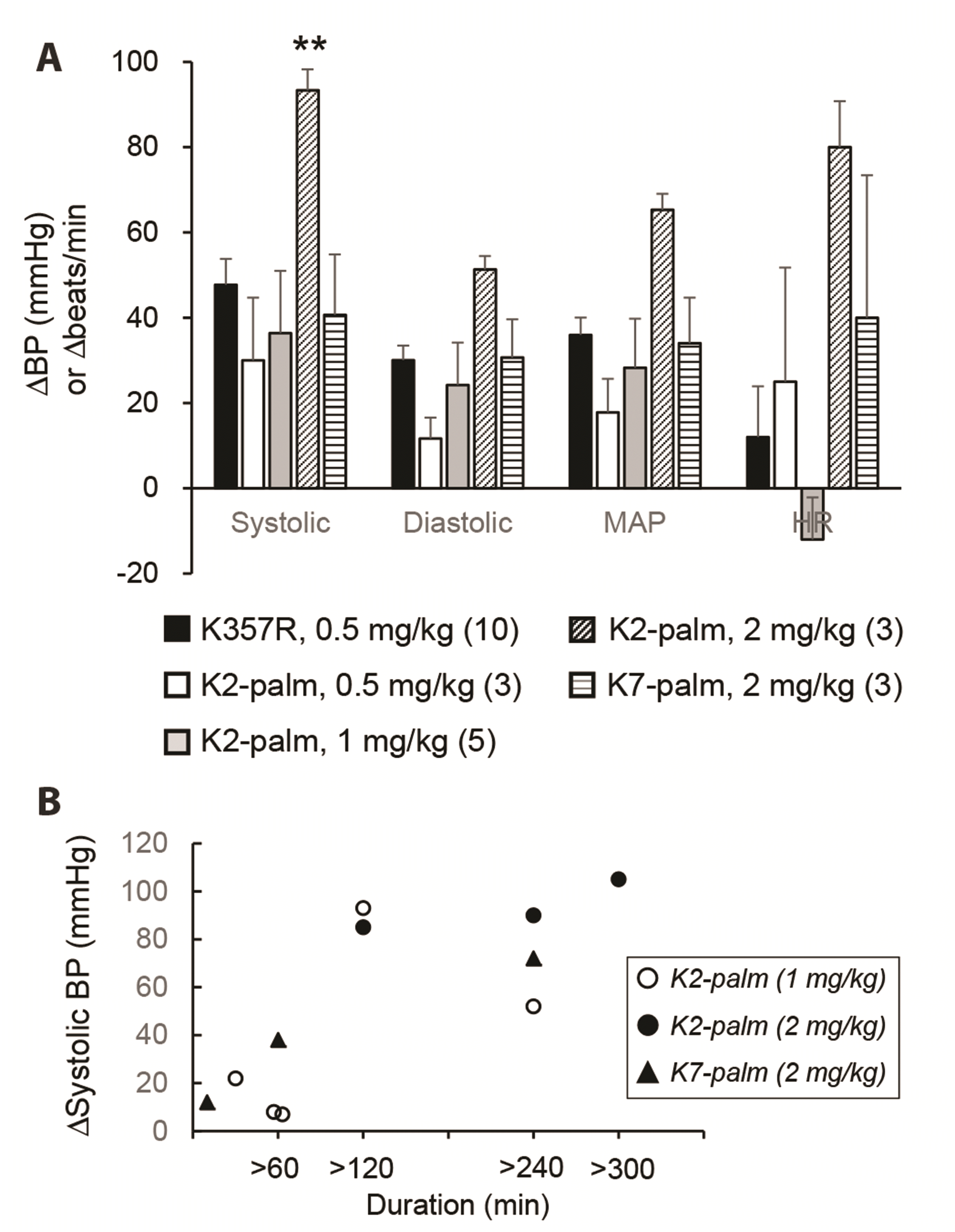 | Fig. 7Prolongation of the blood pressure (BP)-reducing effect by conjugation of palmitic acid with the lysine of cacb1(344–359)K357R.(A) Histogram of the effects of the palmitic acid-conjugated cacb1(344–359)K357R peptide (“K2-palm”, because of the conjugation at the 2nd amino acid, lysine) on systolic, diastolic, and mean arterial pressure (MAP) and heart rate (HR). The BP-reducing effects of K2-palm were dose-dependent, with a significant increase at 2 mg/kg of the systolic pressure (**p < 0.01 vs. K357R). The cacb1(344–359)K357R peptide conjugated with palmitic acid at its 7th residue (K7-palm) at the dose of 2 mg/kg produced an effect similar to that of the K2 palm peptide at the dose of ~0.5–1 mg/kg. Numbers in parentheses indicate the number of rats used. (B) Scatterplot of the duration of the BP-reducing effect by palmitic acid-conjugated peptides with changes in systolic pressure in each rat (K2-palm, 1 mg/kg, open circle; K2-palm, 2 mg/kg, closed circle; K7-palm, 2 mg/kg, closed triangle).
|
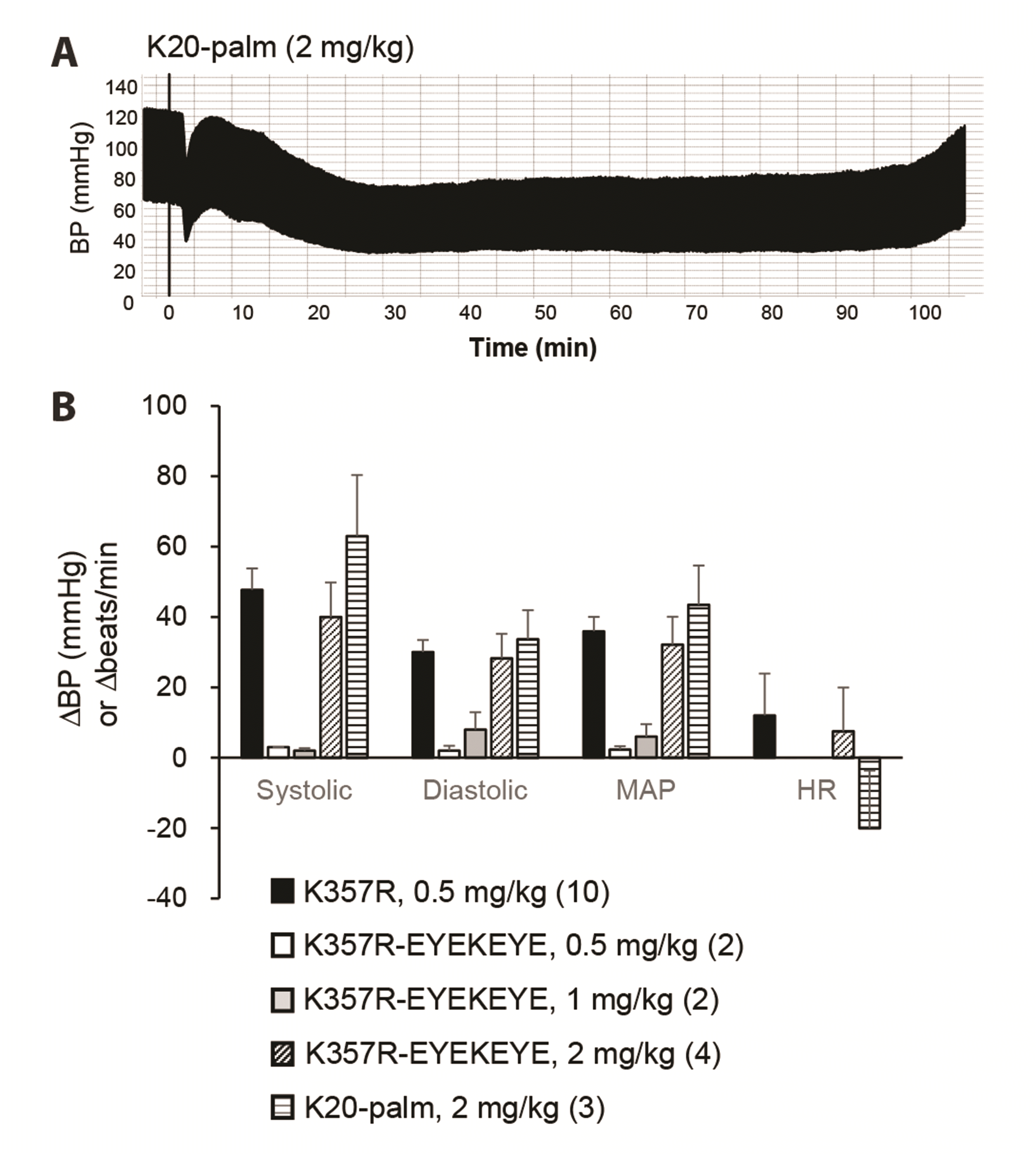 | Fig. 8Effect of the EYEKEYE-added peptide with the conjugation of palmitic acid at the 20th lysine.(A) Intravenous injection of the fusion peptide, cacb1(344–359)K357R-EYEKEYE, with conjugation of palmitic acid at its 20th residue (K20-palm) prolonged the decrease of arterial blood pressure (BP) for at least 90 min. (B) Effect of the fusion peptide cacb1(344–359)K357R-EYEKEYE and the palmitic acid-conjugated fusion peptide (K20-palm) on systolic, diastolic, and mean arterial pressure (MAP) and heart rate (HR). The fusion peptide generated a less potent decrease in BP than the cacb1(344–359)K357R peptide (K357R). Numbers in parentheses indicate the number of rats used.
|
Palmitic acid-conjugated peptide showed prolonged blood pressure-reducing effect in non-anesthetized rats
Because the blood pressure-reducing effect of cacb1(344–359)K357R was significantly prolonged by palmitic acid conjugation at its lysine amino acid (K2-palm), we further tested whether the peptide reduced blood pressure for more than a day. For this, we measured the blood pressure in non-anesthetized rats using the indirect tail-cuff method. In this experiment, K2-palm was further protected from peptidases by N-terminal acetylation, and the acetylated K2-palm peptide maintained its efficacy against systolic, diastolic, and mean arterial pressure (Fig. 9A). When the acetylated K2-palm peptide was administered through the tail vein in non-anesthetized rats, the blood pressure, indirectly measured by tail-cuff, was significantly reduced at 3 h after administration, but after 24 h, it returned to the baseline (Fig. 9B). This indicates that the several-hour blood pressure reducing-effect of K2-palm can be reproduced in non-anesthetized rats.
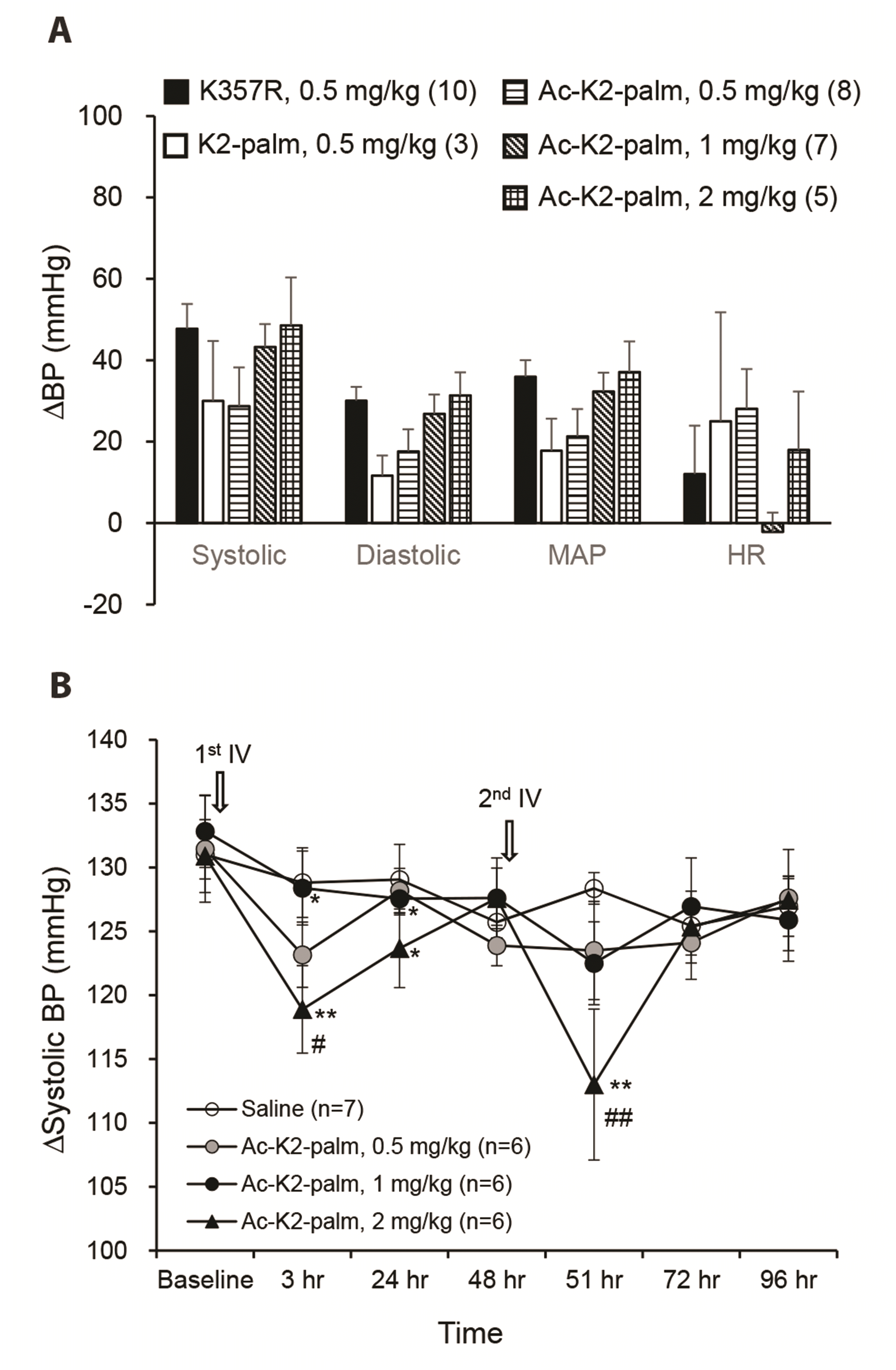 | Fig. 9Effect of the palmitic acid-conjugated cacb1(344–359)K357R peptide on the arterial systolic blood pressure (BP) of non-anesthetized rats.(A) N-terminal acetylation of the palmitic acid-conjugated cacb1(344–359)K357R peptide (K2-palm) did not alter the BP-reducing effect in the anesthetized rats. (B) Intravenous (tail vein) administration (first and second) of the acetylated K2-palm peptide (Ac-K2-palm) decreased systolic BP in non-anesthetized rats recorded by the tail-cuff method. *p < 0.05 and **p < 0.01 vs. before the administration; #p < 0.05 and ##p < 0.01 vs. saline control. Values are presented as mean ± SEM.MAP, mean arterial pressure; HR, heart rate.
|
Acute inflammatory pain was not affected by the HGCC β subunit-derived peptide
Despite of the potent antihypertensive effects of the peptide fragments derived from HGCC auxiliary β subunits, we further tested whether they have any effect on acutely-evoked inflammatory pain. As a result, the intrathecal administration of the Ac-cacb1(344-359)K357R-NH2 peptide (10 µl at 10 mM), which was acetylated at N-terminal and amidated at C-terminal for enzymatic protection, did not change the spontaneous pain behaviors (paw withdrawal and licking) induced by intradermal injection of formalin solution (5%, 50 µl), compared to those induced in the rats administered with the control scrambled peptide (Fig. 10).
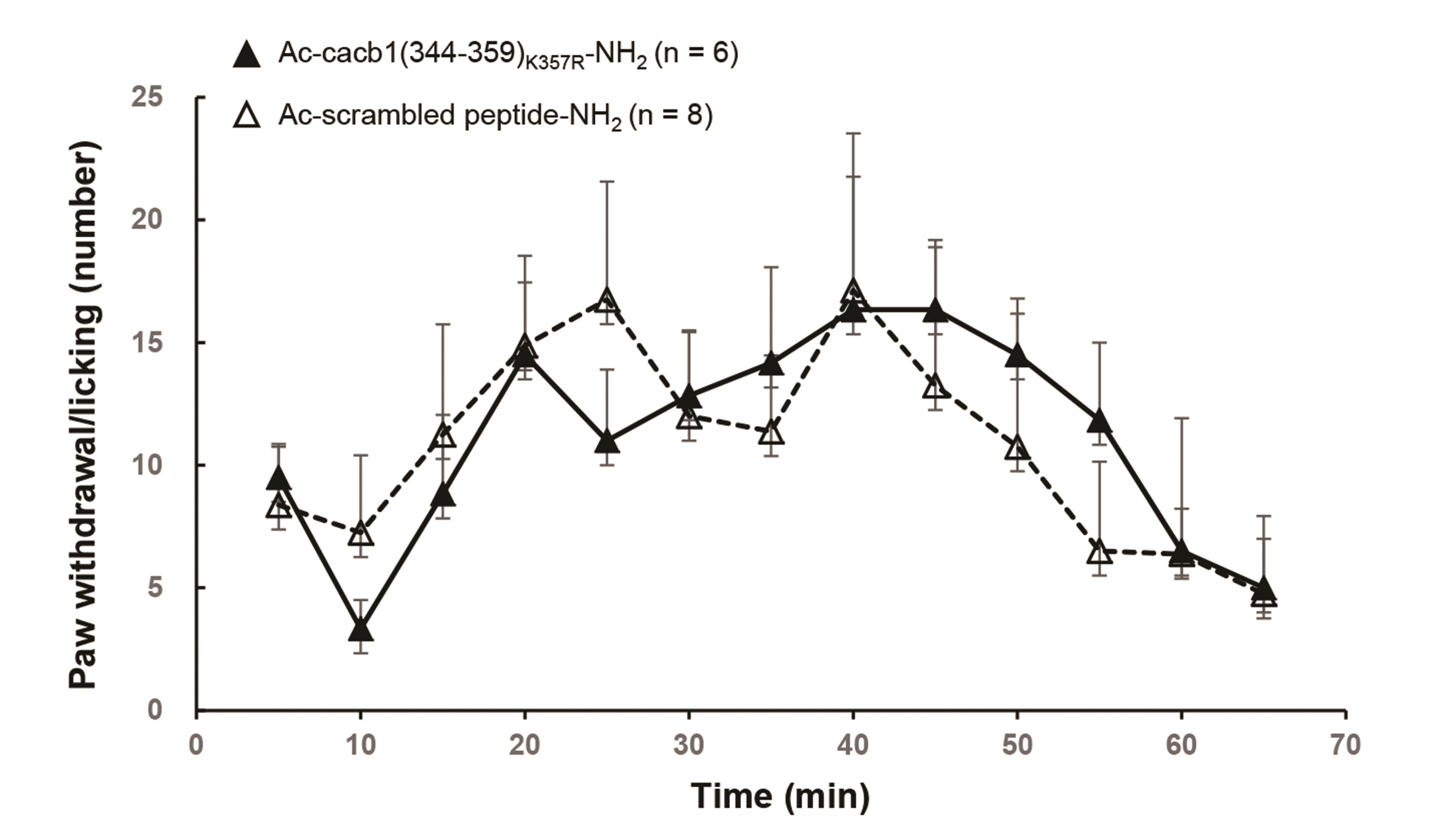 | Fig. 10Effect of Ac-cacb1(344–359)K357R-NH2 peptide on the formalin-induced acute pain.Intrathecal administration of acetylated (Ac) and amidated (NH2) cacb1(344–359)K357R peptide for protection in vivo did not change the pain behaviors induced by intradermal formalin injection (5%, 50 µl) on the plantar surface of rat hindpaw. Values are presented as mean ± SEM.
|
Go to :
DISCUSSION
Intravenous administration of 16-mer peptide fragments [cacb1(344–359)], derived from the same region of HGCC β1, β2, β3, and β4 subunits, robustly decreased arterial blood pressure in anesthetized rats. Although the effect of peptides was not significantly affected by mutations at the four amino acid sites of the peptide, the cacb1(344–359)K357R peptide, whose effect was robust and consistent, was nullified by truncations of two amino acids at either the N-terminal or C-terminal end. In addition, the blood pressure-reducing effect of cacb1(344–359)K357R was prolonged for up to several hours by the conjugation of palmitic acid at its second amino acid, lysine, without losing its potency. This prolonged effect was also observed in non-anesthetized rats.
In a previous study, we showed that two different peptides, VT and LD, were derived from the α3 helix and β10 sheet-α9 helix parts of the AID-binding sequence in the β subunit [5], reducing HGCC functions in acutely isolated TGs [13]. However, we found no changes in the peak amplitudes of voltage-activated Ca2+ current in the TG neurons by applying the peptides derived from the β9 sheet-α6 helix region of β subunits internally through patch pipettes. Referring to the structural basis of α subunit interactions, the interacting regions in the AID of the α subunit are slightly different among the α3, α6, and α9 helices in the β subunit [5]. The α6 helix interacts with the N-terminal amino acid leucine, in addition to the main hot spot amino acids tyrosine, tryptophan, and isoleucine, in the AID, whereas the α3 and α9 helices interact with the backward amino acids glutamine and alanine [5]. This difference may affect the affinity of the peptide fragments to the binding region of the α subunit, thereby altering the effectiveness in the function of HGCCs. Otherwise, the peptide fragments possibly affected a specific subfamily of HGCCs, which contributed little to the total voltage-activated Ca2+ current.
The length of peptide fragments (i.e., 16 amino acids) was essential for the effect on blood pressure, as the efficacy disappeared by truncation at the N- or C-terminals. Regarding the amino acids in the interaction between the β subunit and the AID of α subunit, the first amino acid at the N-terminal and the last amino acid at the C-terminal—isoleucine and arginine (Table 1), respectively—are essential interacting residues in the peptide fragment [5]. Therefore, it is suitable that their truncation nullifies the effect of the peptide.
In this study, the blood pressure-reducing effect of 16-mer peptides was strong but short-lasting; thus, we attempted to prolong this effect by increasing their plasma half-life. Generally, conjugating small peptide drugs to ligands that bind large serum proteins prevents fast glomerular filtration, thereby prolonging drug efficacy through the extension of plasma half-life [14]. Thus, we conjugated palmitic acid, an albumin ligand [15,16], to lysine at the 2nd or 7th amino acid position of the cacb1(344–359)K357R peptide. Surprisingly, these conjugated peptides substantially prolonged the effects on arterial blood pressure (> 4 h), although their potency was slightly different (K2-palm > K7-palm). On the other hand, certain peptide sequences surrounding palmitic acid-bound lysine improve the binding capacity of palmitic acid to albumin [17]. Therefore, we added the peptide sequence EYEKEYE to cacb1(344–359)K357R; then, palmitic acid was conjugated with the lysine of the attached residue (K20-palm). This peptide also showed a prolonged blood pressure-reducing effect but was less potent than those of K2-palm and K7-palm. Therefore, we concluded that palmitic acid conjugation at the first lysine (second residue) of cacb1(344-359)K357R was the best modification for prolonging the blood pressure-reducing effect.
As the peptides derived from the β subunit of HGCCs exert a blood pressure-reducing effect by intravenous administration but do not reduce whole-cell Ca2+ currents by intracellular administration, the peptides may interact with many known molecular targets for hypertension, such as angiotensin I converting enzyme (ACE), angiotensin II type I receptor (AT1R), endothelin receptors, or nitric oxide donors [18], which are examples from studies using peptides derived from food, particularly casein, a major protein in milk [19-21]. Many small- or medium-sized peptides from casein showed inhibitory activity against ACE in vitro [20], or were absorbed into the bloodstream upon oral administration and reduced systolic blood pressure, potentially via inhibition of the AT1R [19]. This suggests that a certain peptide fragment exerts a hypertensive effect if it binds or docks to an optimal site at the molecular level and exists as an antihypertensive target, which is consistent with hypertension being caused by various molecular abnormalities in the cardiovascular or nervous system; thus, a molecule may exert antihypertensive effects if it manipulates the function of any target. Therefore, the potential molecular targets, except for the HGCCs, of the peptides derived from the β subunit must be investigated.
In this study, we report for the first time that 16-mer peptides from the β subunits of HGCCs have potent antihypertensive effects in anesthetized and non-anesthetized rats, without any effect on acute pain responses. These results were not expected because these peptides do not cross cell membranes nor interfere with the α-β interactions of HGCCs. Therefore, the mechanism of the blood pressure-reducing effect of these peptides was not identified. However, many drugs are accidentally discovered and then marketed without knowledge of their pharmacodynamic mechanisms [22]; therefore, we expect the 16-mer peptides herein reported to be valuable for the treatment of hypertension.
Despite the difficulty in crossing cell membranes, peptide drugs are generally advantaged compared to small-molecule drugs because their larger surface area for binding and chiral complexity allow for better selectivity, which lowers the likelihood of off-target or side effects [22]. In addition, peptide drugs can be easily modified into chemically stable structures and used in molecular techniques such as the introduction of the genetic code for the peptide sequence into implantable stem cells. Thus, the peptide fragments herein shown may be interesting for usage in blood pressure control, considering the shortage of peptide drugs for antihypertensives [23]. Furthermore, the invention of solid-phase peptide synthesis [24] has lowered the overall costs of peptide drug development compared to those of small molecules because of their intrinsic synthetic feasibility, which leads to a shorter period for lead optimization [22]. With this, the approval of peptide drugs has steadily increased over the last six decades, with a global pharmaceutical market proportion of 5% in 2019 [22]. The clear blood pressure-reducing effect of our peptides may result in their use as the next generation of antihypertensive drugs if further drug developmental processes are successful.
In summary, we report here for the first time that peptide fragments from β subunits of HGCCs have potent blood pressure-reducing effects in anaesthetized and non-anaesthetized rats. The effect was crippled by only two-amino acid truncation at N-terminal or C-terminal end, and was tremendously prolonged by conjugating palmitic acid through the second amino acid, lysine, without losing its potency. Considering the shortage of peptide drugs for antihypertensives, we expect that this finding provides a new tool for treating hypertension.
Go to :
 XML Download
XML Download