

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
Y-family DNA polymerases (Polη, Polτ, Polκ, and Rev1) specialize in translesion DNA synthesis (TLS), facilitating cells with damaged DNA to continue DNA replication [1,2]. The B-family TLS DNA polymerase zeta (Polζ) consists of two subunits, Rev3 and Rev7, and plays a critical role in translesion bypass replication in most eukaryotes, including humans [3]. In response to most types of DNA damage, the complex of Polζ and Y-family polymerase Rev1 is recruited to the damaged sites and participates in error-prone DNA repair [4]. Central to lesion bypass by Polζ and Rev1 is the recruitment of the E3 ubiquitin ligase Rad6 (E2)-Rad18 (E3) to the site of DNA damage, where it ubiquitinates the proliferating cell nuclear antigen (PCNA) [5]. Y-family polymerases have ubiquitin-binding domains that bind to monoubiquitylated PCNA [4].
Rev7 is identical to the mitotic arrest deficient 2 like 2 (Mad2L2, also known as Mad2B) protein in terms of its amino acid sequence [6]. Mad2B shares high sequence homology with Mad2, a spindle assembly checkpoint (SAC) protein [7], suggesting that Mad2B also functions as a SAC protein [8,9]. The main effector of SAC is the mitotic checkpoint complex, comprising BubR1, Bub3, and Mad2, which together bind to cell division cycle (Cdc)-20, a co-activator of the anaphase-promoting complex or cyclosome (APC/C), and E3 ubiquitin ligase [10]. Mad2B also binds to Cdh1 and/or Cdc20 to impede APC activation [8,9].
Numerous studies have identified the critical role for Mad2B in the DNA damage response (DDR) pathway in human cells [11-14]. Mad2B is recruited to DNA double-strand breaks and participates in non-homologous end-joining (NHEJ) repair for DNA damage tolerance via the Polζ function [13]. Mad2B regulates DNA repair by 53BP1, which promotes NHEJ [11]. DNA damage initiates DDR that ultimately results in the recruitment of repair proteins to the sites of DNA damage [15]. Current models [16,17] describing the mechanism of cisplatin-induced DNA damage indicate that many proteins, including endonucleases and DNA repair polymerases, are recruited to the sites of DNA damage. Mad2B also associates with various proteins depending on the cell requirement; it binds to Cdh1, RAN GTPase, and clathrin light chain A for mitotic regulation, and to Rev3, Rev1, POLD2, 53BP1, and SHLDs for TLS activity or DNA damage repair [18].
As our previous study has implicated Mad2B/Rev7 in DDR [19], we have evaluated the mechanism by which Mad2B functions during cisplatin-induced DNA damage. In this study, we investigated precisely what role Mad2B plays in APC/C activity in binding to Cdc27. So far, Mad2B is known to bind to Cdc27 only in mitosis to inhibit APC/C activity [20], but its binding during DDR remains unknown. Here we further show, in human cells, that Mad2B/Rev7 exists as a Polζ-Rev1-Cdc27 complex in undamaged cells, with low ubiquitylation activity, which recruits Cdc20 in response to DNA damage, thereby increasing APC/C activity to ubiquitylate target proteins. Our data identify a previously unknown role for Mad2B-Cdc20-APC/C in DDR.
METHODS
Cell culture and preparation of cell extracts
Human cervical carcinoma cells (HeLa) and HEK293 cells were obtained from BioWhittaker Europe and cultured as described previously [21]. HeLa cell was lysed in either buffer A or RIPA buffer (Thermo Scientific). Cell lysates were centrifuged at 14,000 g for 10 min at 4°C and the protein content of the supernatant determined using the Coomassie Protein Assay Reagent (Thermo Scientific) before normalisation and solubilisation in x2 SDS-PAGE sample buffer. Soluble and chromatin protein fractions were prepared as described previously [22].
Cell proliferation assay
Cell proliferation was estimated using a colorimetric method based on water-soluble tetrazolium salts (WST-1) (CellVia; AbFrontier) according to the manufacturer’s instructions. Briefly, 1.5 × 104 cells/well were placed in a 96-well microplate and incubated overnight. Cells were then treated with cisplatin at different concentrations and for varying times. After cisplatin addition, 10 μl of WST-1 reagent was added to 100 μl of culture medium in each well, and incubation was continued for 2 h. The absorbance of the samples was measured at 440 nm using a 96-well microplate spectrophotometer (Multiskan GO; Thermo Fisher Scientific).
DNA damage
To induce double strand DNA damage exponentially growing HeLa cells were treated with cisplatin (50–100 μM) for varying times.
Plasmids
The human Mad2B gene was cloned by PCR amplification from a human HeLa cell cDNA library (Clontech) using sequence-specific primers. One set of primers was also designed to incorporate an HA-epitope tag at the 5’ terminus of the Mad2B gene. The non-epitope tagged genes were subcloned into pGEX-4T-1 (Amersham-Pharmacia) and the HA-epitope tagged Mad2B gene was subcloned into a mammalian expression vector (pCMV5). The pEF-4myc-Cdc20 expression vector was generated by cloning full length human Cdc20 downstream of 4 myc tags under an elongation factor 1 promoter. hRev1 and hRev3 truncation mutants were generated by PCR amplication of full-length hRev3 and hRev1. For generation of expression vectors the amplicons of hRev1 (aa 907-1251 containing the Mad2B binding region and aa 1-343, the non-Mad2B-binding region) and hRev3 (aa 1665-2070 containing the Mad2B binding region and aa 2680-3130, the non-Mad2B-binding region) were subcloned into the pLEICS12 vector by Protein Expression Laboratory (Protex, University of Leicester). Expression from this vector produces N-terminally Flag-tagged protein.
Antibodies
The following antibodies were purchased from Santa Cruz: monoclonal anti-HA, rabbit polyclonal anti-HA and polyclonal anti-Cdc20. Rabbit polyclonal antibodies to Histone H3, Cdc27 and PHistone-H2AX (Ser139) antibodies were purchased from Cell Signalling Technologies. The following antibodies were purchased from Sigma-Aldrich: monoclonal anti-g-tubulin, polyclonal α-tubulin, HRP-conjugated goat anti-mouse, HRP-conjugated goat anti-rabbit. A human anti-centromere antibody was purchased from Europa Bioproducts Ltd. Monoclonal antibodies to PCNA, Mad2B and c-myc were purchased from BD Transduction Laboratories. The following antibodies were purchased from Invitrogen: Alexa Fluor 488 goat anti-mouse IgG, Alexa Fluor 488 goat anti-rabbit IgG, Alexa Fluor 594 rabbit anti-mouse IgG and Alexa Fluor 594 goat anti-human IgG.
Transfection of HeLa cells
HeLa cells were transfected using Fugene 6 (Roche) as described previously [21].
RNAi
The Mad2B oligonucleotides and non-targeting, control small interfering RNA (siRNA) oligonucleotide was purchased from Dharmacon Research Inc. The sequence of the sense strand of the siRNA duplexes were as follows: Mad2B, 5’-CCAAAGUUGAGGUCUUGUCUU-3’, Control, 5’-UAGCGACUAAACACAUCAA-3’. The siRNA oligonucleotides were dissolved in RNAse-free buffer (20 mM KCl, 6 mM Hepes, 0.2 mM MgCl2, pH 7.5) at a concentration of 20 μM. Cells were transfected with the siRNA oligonucleotides using Interferin transfection reagent (Polyplus) according to the manufacturers protocol.
Immunoprecipitation (IP) and Western blot analysis
IP were performed as described previously [21]. In some experiments the immunoprecipitates were not heated in order to minimise degradation of the IgG. The immunoprecipitated proteins and the cell extracts were resolved by SDS-PAGE and electroblotted onto Hybond-C nitrocellulose membrane (Amersham-Pharmacia) using a Hoeffer semi-dry blotting apparatus (Amersham-Pharmacia Biotech). Immunoreactive proteins were visualised using enhanced chemiluminescence according to the manufacturer’s instructions (Geneflow).
Purification of recombinant glutathione-S-transferase (GST) fusion protein
Recombinant GST-Mad2B was prepared as described previously [23]. Fast protein liquid chromatography (FPLC) purification was performed using an ÄKTA system (Amersham). GST-tagged Mad2B protein were purified by passing them twice through a glutathione-agarose column (Sigma). To further remove minor contaminating proteins from the Mad2B fractions both ion exchange and gel filtration (Superdex 75; GE Healthcare) chromatography were performed.
GST-pulldown assay
To examine the interaction between Cdc20 and Mad2B interphase, HeLa cells following cisplatin treatment were lysed in buffer A and incubated (4ºC for 1 h) with purified, recombinant GST-Mad2B or GST at varying concentrations (between 0.2–1.0 μM). Glutathione-Sepharose beads (10 μl packed volume, GE Healthcare) were added to each tube and incubated at 4ºC for 1 h. The beads were washed 3 times in cold (4ºC) Dulbecco’s PBS and the bound Cdc20 protein analysed by immunoblotting with a polyclonal Cdc20 antibody.
Immunofluorescence microscopy
Cells were fixed and processed for immunofluorescence microscopy as described previously [21]. Where indicated cells were pre-extracted with 0.1% v/v Triton X-100 in Dulbecco’s PBS for 2–5 min prior to fixation and immunostaining.
In vitro ubiquitination assay
The in vitro ubiquitination assays were performed as described previously [24].
Reagents
All other reagents were of analytical grade and obtained from either Sigma-Aldrich or Fisher.
Statistical analysis
All data are presented as the mean ± SD. The n-value represents the number of independent samples used in the experiments. The significance for all pairwise comparisons of interest was assessed using the two-tailed Student’s t-test, and p < 0.05 was considered statistically significant. All experiments were independently performed with at least three different samples. GraphPad Prism 4 software (GraphPad Software) was used for statistical analysis.
RESULTS
Mad2B levels increase in HeLa cell after cisplatin-induced DNA damage
We confirmed that the activation of PCNA and phospho-H2A.X, DNA damage markers, which occurred following cisplatin treatment, induced DNA damage. HeLa cells were treated with 100 μM cisplatin for 12 h, followed by staining with anti-PCNA and anti-pH2A. X antibodies (S139). As shown in Fig. 1A and B, treatment with cisplatin DNA damaging agent induced the activation of PCNA and phosphorylation of H2A.X. Positive staining was observed in approximately 80% of the cells. These results indicate that cisplatin treatment induces DNA damage in HeLa cells.
We examined whether Mad2B was also recruited to DNA damage sites following cisplatin treatment in HeLa cells. Western blotting analysis of cytoplasmic and chromatin-associated proteins indicated that Mad2B was primarily a chromatin-associated protein (Fig. 1C). To show that Mad2B expression levels were altered following cisplatin treatment, HeLa cells were collected at different intervals following cisplatin treatment. The results of Western blotting indicated that there was a gradual increase in the levels of Mad2B protein following cisplatin addition in a time-dependent manner (Fig. 1D).
Mad2B binds to Cdc20 following DNA damage
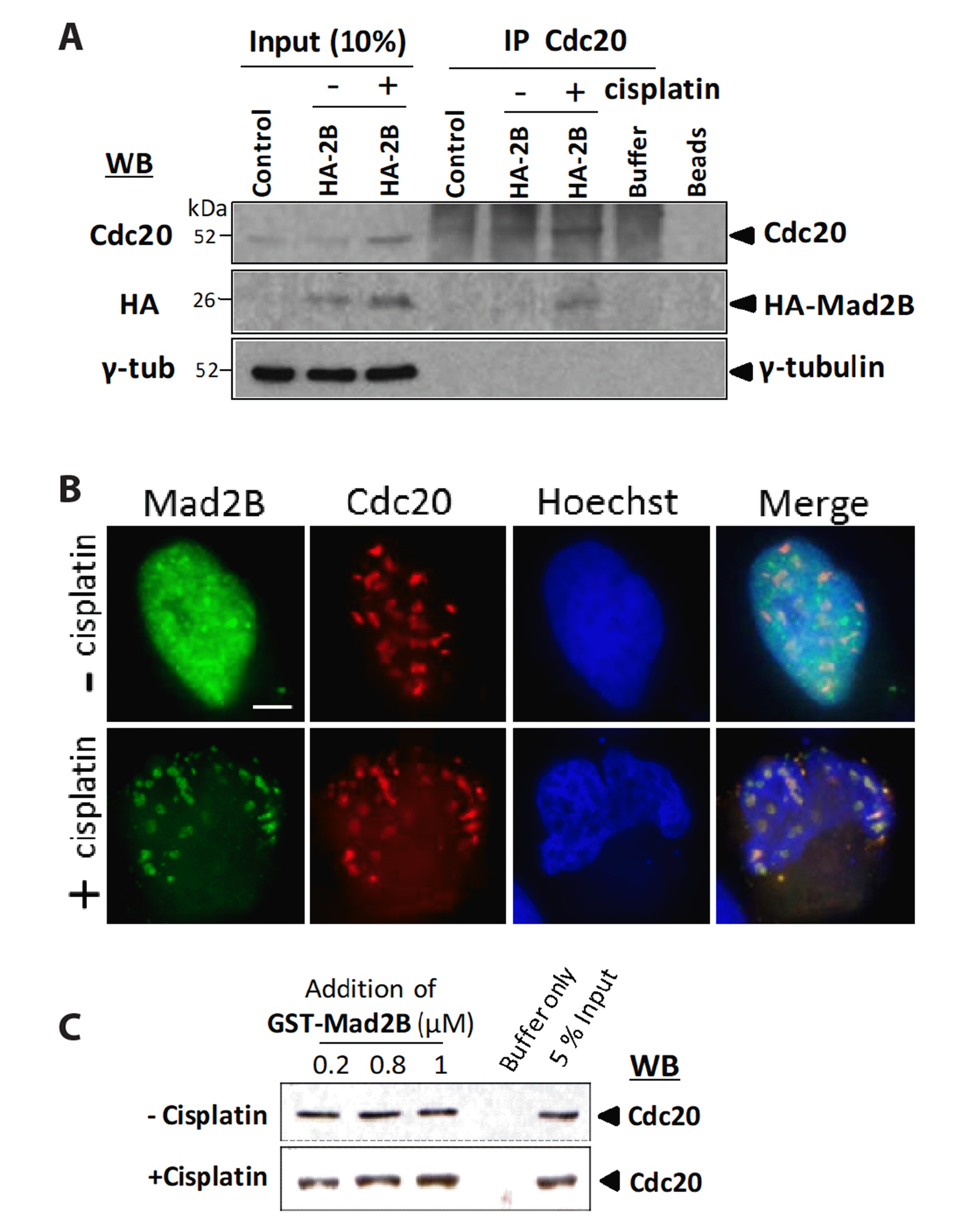
To determine whether Mad2B bound to Cdc20 following the activation of the DNA damage checkpoint, we performed IP of endogenous Cdc20 from HeLa cells overexpressing HA-Mad2B. Immunoblotting analysis of Cdc20 revealed that Mad2B co-immunoprecipitated with Cdc20 following cisplatin treatment although only at a low level (Fig. 2A). To further confirm the interaction between Mad2B and Cdc20, HeLa cells were co-transfected with Myc-Cdc20 and HA-Mad2B. Examination of cells co-expressing HA-Mad2B and Myc-Cdc20 via immunofluorescence microscopy indicated that both proteins colocalized in the nucleus only after cisplatin treatment (Fig. 2B). To determine whether the recombinant Mad2B could interact directly with native Cdc20 in cisplatin-treated HeLa cells, we performed a GST-pulldown assay using recombinant GST-Mad2B (0.2–1 μM). HeLa cell lysates were prepared from exponentially growing cells (interphase) and cells treated with cisplatin (50 μM) for 18 h. A Western blot analysis of proteins bound to the glutathione-sepharose beads indicated that Mad2B (0.2–1 μM) displayed a uniform level of binding to Cdc20. However, GST-Mad2B-bound Cdc20 increased in a concentration-dependent manner in the cisplatin-treated cells (Fig. 2C). These results indicated that Mad2B was capable of specifically binding to Cdc20 in HeLa cells in vitro following cisplatin-induced DNA damage (Fig. 2C). These data suggest that Mad2B binds to Cdc20 following cisplatin-induced DNA damage.
Mad2B, Rev3, and Rev1 complexes associate with the APC/C subunit Cdc27
As Mad2B recruits the APC/C co-activator, Cdc20, following DNA damage, we examined whether Mad2B was also associated with the APC/C. We performed co-IP to assess whether Mad2B bound to Cdc27 (also known as APC3), a subunit of the APC/C. Endogenous Cdc27 was immunoprecipitated from HeLa cells, both before and after cisplatin treatment (50 μM for 18 h). The results (Fig. 3A) indicated that native Mad2B was associated with endogenous Cdc27 both before and after cisplatin treatment. However, the level of Mad2B associated with Cdc27 increased following cisplatin treatment (Fig. 3A, B). Further confirmation of the interaction between Mad2B and Cdc27 was obtained by transfecting HEK293 cells with HA-Mad2B. As shown in Fig. 3B, immunoblotting of the Cdc27 immunoprecipitates indicated that Ha-Mad2B was associated with Cdc27 prior to cisplatin treatment. Additionally, the amount of Mad2B that associated with Cdc27 increased after cisplatin treatment (Fig. 3B). To further confirm the association between Mad2B and Cdc27, we determined the intracellular localization of HA-Mad2B and Cdc27, both before and after cisplatin treatment, using immunofluorescence microscopy. The results (Fig. 3C) indicated that HA-Mad2B co-localized with native Cdc27 in the nuclei of HeLa cells, both before and after cisplatin treatment (Fig. 3C). However, cisplatin altered the distribution of Cdc27 and HA-Mad2B from a particulate to a more a more finer punctate distribution.
As Mad2B/Rev7 is involved in TLS together with its partners, Rev3 and Rev1 [3], it was important to determine whether Mad2B binds to Cdc27 independently of Rev3 and Rev1. Because both hRev1 and hRev3 are large proteins, we generated N-terminal FLAG-tagged deletion constructs for each protein. HEK293 cells were co-transfected with HA-Mad2B and either a FLAG-tagged fragment of hRev1 (aa. 907–1251 containing the binding domain for Mad2B/Rev70) [25,26] or a FLAG-tagged fragment of hRev3 (aa. 1665–2070 containing the binding region for Mad2B/Rev7) [25]. Western blotting analysis of HA-Mad2B immunoprecipitates following cisplatin treatment indicated that Mad2B was constitutively associated with the binding fragments of hRev1 (Fig. 4A) and hRev3 (Fig. 4B), both before and after cisplatin treatment. In parallel experiments, performed using the non-Mad2B/Rev7-binding fragment of hRev1 (a.a. 1–343) or hRev3 (a.a. 2680–3130), no association of either hRev1 or hRev3 fragment with HA-Mad2B was observed (Supplementary Fig. 1). To further investigate the interaction between the Mad2B/hRev1/hRev3 complex and Cdc27, we performed a co-IP experiment. Native Cdc27 was immunoprecipitated from HEK293 cells that been co-transfected with HA-Mad2B and either FLAG-hRev1 (a.a. 907–1251) or FLAG-hRev3 (a.a. 1665–2070) and then treated with cisplatin (50 μM for 18 h). Western blot analysis of the Cdc27 immunoprecipitates indicated that the Mad2B/hRev1 complex was associated with Cdc27 prior to cisplatin treatment, but the recruitment of the Mad2B/hRev1 complex to Cdc27 increased after cisplatin treatment (Fig. 4C). A similar increase in the recruitment of the Mad2B/hRev3 complex to Cdc27 was also observed following cisplatin treatment (Fig. 4D). These results suggest the existence of a protein complex in the nuclei of cells, comprising the translesion DNA repair proteins, Mad2B (hRev7), hRev1, hRev3, and the APC/C component, Cdc27.
Additional recruitment of Mad2B to hRev1, hRev3 and Cdc27 occurs following cisplatin-induced DNA damage.
APC/C activity is enhanced in the presence of Mad2B following cisplatin-induced DNA damage
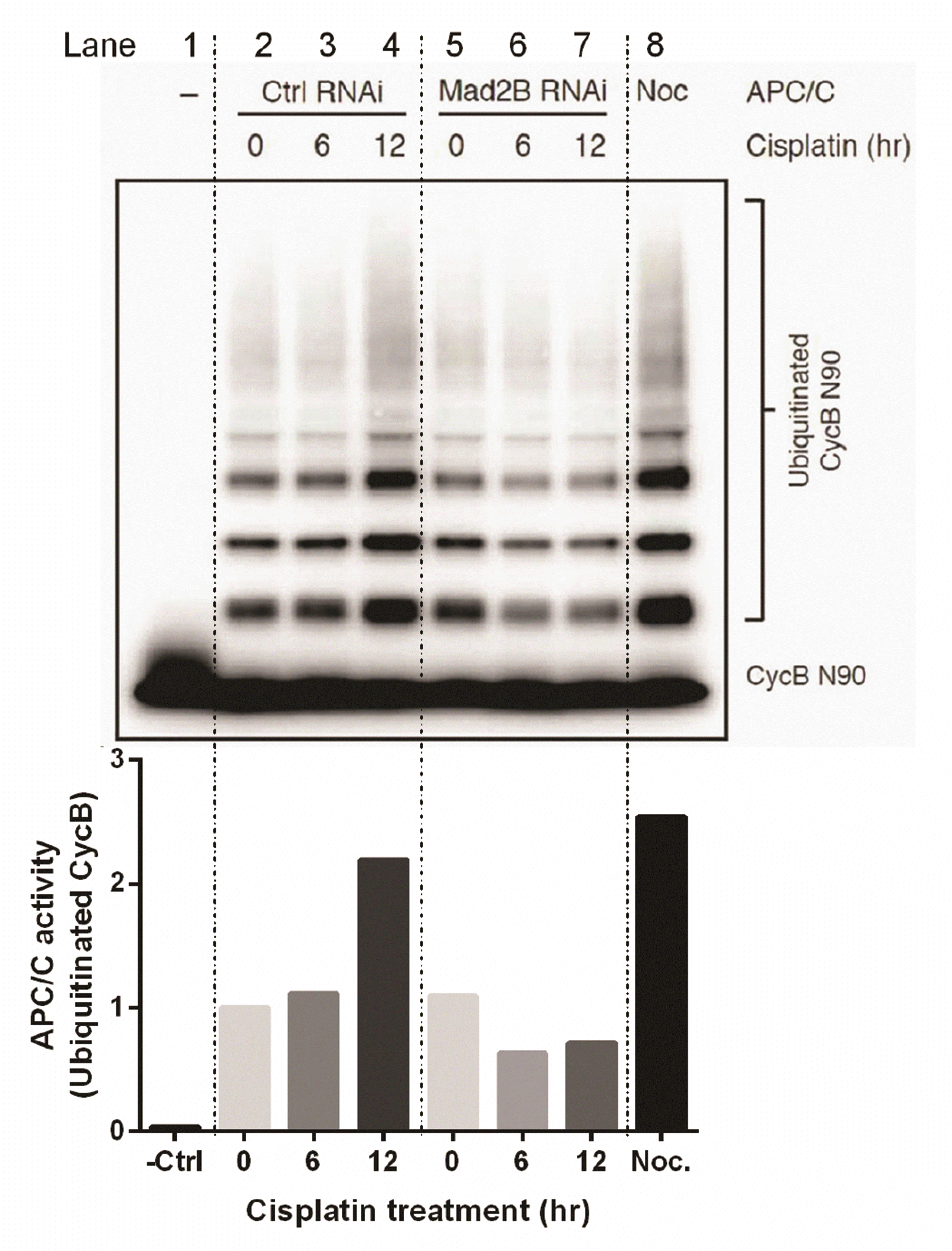
Our results have demonstrated that Mad2B binds to the APC/C subunit, Cdc27, and that its binding to Cdcd27 is increased in the presence of DNA damage. Therefore, we investigated whether Mad2B affected the activity of APC/C ubiquitination in response to DNA damage. We analysed APC/C activity in the absence or presence of Mad2B in cisplatin-induced DNA damage using an in vitro ubiquitination assay. HeLa cells were transfected with the Mad2B siRNA (10 nM). Then, 24 h post post-transfection, the cells were treated with 50 μM cisplatin for various time periods (0, 6, and 12 h). Ubiquitinated cyclin B was analyzed via Western blotting using an anti-Myc antibody. The results of the in vitro ubiquitination assay revealed that APC/C ubiquitinated cyclin B in the presence of Mad2B and its activity was increased more than 2-fold in HeLa cells in response to cisplatin-induced DNA damage in a time-dependent manner (lanes 2, 3, and 4 in Fig. 5). However, in the absence of Mad2B, the APC/C ubiquitylation activity for cyclin B was reduced by almost half in HeLa cells after DNA damage (lanes 5, 6, and 7 in Fig. 5). These data indicate that Mad2B may act as an activator of APC/C ubiquitination in human cancer cells following cisplatin-induced DNA damage.
DISCUSSION
Initial studies of Mad2B suggested that it may function in the SAC [8,9,27], but various other studies have shown that Mad2B is involved in the DDR [11-14,28]. Mad2B (also known as Rev7), together with Rev3 (called Polζ) and Rev1, is involved in mutagenic TLS [3,12,29]. The findings of the current study are consistent with the role of Mad2B in DNA damage and repair.
However, in this study, we have extended understanding of the mechanism of Mad2B during DDR. We showed Mad2B recruits both Cdc27 (APC3, a TPR-containing subunit of the APC/C) [10,30] and Cdc20 (a co-activator of the APC/C) [31] following cisplatin-induced DNA damage. Our data suggests the involvement of Mad2B-Cdc20-APC/C in the cisplatin-mediated DDR.
The APC/C is a large protein complex that plays a crucial role in regulating the cell cycle by targeting specific proteins for degradation [10]. One of the key regulators of APC/C activity is Mad2B, which has been shown to inhibit APC/C function by binding to Cdc27 specifically during mitosis [20]. However, we showed that Mad2B binding to Cdc27 and Cdc20 occurs in the DDR in cisplatin-treated cells, and that this binding conversely enhances the activity of APC/C.
Cisplatin treatment in HeLa cells induced the activation of PCNA and phosphorylation of γH2AX (Fig. 1A, B), which are markers of DNA damage [32,33]. This confirmed that cisplatin treatment triggered DNA damage in cells. The levels of Mad2B, a chromatin-associated protein, were gradually increased in human cancer cells after cisplatin treatment (Fig. 1C, D). This suggests that Mad2B is recruited to the site of DNA damage in cancer cells after cisplatin-induced DNA damage. As shown in previous results, treatment with 50 uM cisplatin induced DNA damage indicating phosphorylation of Chk1, leading to apoptosis in HeLa cells [19]. In addition, present study indicated that 50 uM cisplatin addition showed 80% cell viability in HeLa cells (Supplementary Fig. 2). Therefore, in this study, this concentration of cisplatin was used to induce DNA damage in cells.
Rev7 can simultaneously interact with both Rev3 and Rev1 [26]. The binding data in our study confirmed the association of Rev7/Mad2B with Rev1- and Rev3-binding fragments (Fig. 4A, B). However, our co-IP data demonstrated the existence of a larger protein complex in undamaged cells, comprising Polζ-Rev1 and Cdc27 (Fig. 4C), but we also newly observed an increase in Mad2B, Rev3, and Rev1 protein recruitment to Cdc27 after cisplatin-induced DNA damage (Fig. 4C, D). Our colocalization experiments clearly support the association between Cdc27 and Rev7/Mad2B in both untreated and cisplatin-treated cells (Fig. 3C). However, cisplatin altered the distribution of both Cdc27 and HA-Mad2B from a particulate to a more punctate phenotype. This was probably due to the generation of DNA damage foci following cisplatin treatment. Furthermore, our observations indicate that Rev7/Mad2B, presumably in association with Rev1, Rev3, and Cdc27 (Fig. 4), binds to Cdc20 (Fig. 2) following DNA damage. The association of Cdc27 (presumably the entire APC/C complex) with Polζ-Rev1 and the additional recruitment of Cdc20 to this complex by Rev7/Mad2B in response to DNA damage has not previously been reported. However, the mechanism by which the complex is assembled and recruited to the sites of DNA damage as well as its function remain unclear.
Following cisplatin-induced DNA damage, additional Cdc20 was recruited to the ternary complex by Rev7/Mad2B (Fig. 2). Our biochemical and colocalization data support the interaction of Rev7/Mad2B following DNA damage. Based on the finding that the binding of Rev7/Mad2B to Cdc20 does not inhibit the activity of APC/C [34,35], we suggest that the additional recruitment of Cdc20 to the ternary complex is consistent with its established role in activating the APC/C. Furthermore, we confirmed that Mad2B affects APC/C ubiquitination in response to DNA damage (Fig. 5). The in vitro ubiquitylation assay indicated that, in the presence of Mad2B, APC/C activity was increased in a time-dependent manner in human cancer cells after cisplatin-induced DNA damage (Fig. 5). This suggests that Mad2B may also act as an activator of APC/C ubiquitination in human cancer cells following cisplatin-induced DNA damage. Cdc20 binds to Cdc27 (APC3) to form a destruction box (D-box) degron receptor that recognizes D-box-containing proteins that are targeted for degradation [36,37]. While our findings in the current study suggest the role of Cdc20-APC/C in the ubiquitination of protein(s) following DNA damage, the identity of the target protein(s) remains unknown. APC/C-Cdc20-mediated ubiquitination of its target(s) may be involved in the recruitment of additional proteins to the DNA damage foci [16] or alternatively in the termination and/or disassembly of the DNA repair complex. A role for the APC/C in DDR has been reported previously [38]. The DNA damage mediator protein, mediator of DNA damage checkpoint 1 (MDC1), directly binds to the Cdc27 (APC 3) subunit of APC/C following double-strand DNA breaks induced by ionizing radiation, although the function of this interaction remains unclear [38]. Moreover, APC/C-Cdh1-mediated ubiquitination and destruction of Rad17, a loader of the 9-1-1 complex, is required to terminate checkpoint signaling in response to UV-induced DNA damage [39]. Mad2B combines with the APC/C subunit, Cdc27, only in the G2/M phase to inhibit APC/C activity [20], and our results newly indicated that Mad2B binds to Cdc27 in the presence of DNA damage.
Chun et al. [40] have reported that overexpression of Mad2B/Rev7 increased Rev1 degradation mediated by APC/C ubiquitination, suggesting an interplay between APC/C function and TLS [40]. Our results showed that Mad2B (Fig. 1C, D) and Cdc20 (Fig. 2A, C) protein levels increased after cisplatin-induced DNA damage. Importantly, the binding of Rev1 and Mad2B to Cdc27 increased after DNA damage (Fig. 4C); thus, Rev1 was not subjected to APC/C-mediated degradation during DDR. This is inconsistent with the results of Chun et al. [40], who showed that APC/C activity was enhanced in the presence of Mad2B in cisplatin-induced DNA damage (Fig. 5).
The amount of Mad2B bound to Cdc27 consistently increased after cisplatin-induced DNA damage. This suggests that Mad2B-bound Cdc27 recruits both APC/C and Cdc20 following cisplatin-induced DNA damage to ubiquitinate target proteins. Binding of Rev3 and Rev1 to Mad2B also increased following cisplatin treatment. Thus, the Rev3/Rev1/Mad2B-APC/CCdc20 complex may be involved in the regulation of DNA damage signaling pathways and may potentially lead to ubiquitination-mediated proteolysis of DNA damage checkpoint or DNA repair proteins. Hence, further analysis of APC/C, its interacting partners, and substrates is required to dissect its specific roles in translesion DNA damage repair.
In summary, during the activation of DNA damage checkpoints after cisplatin treatment, DNA polymerase ζ (Mad2B/Rev7 and Rev3) forms a complex with Rev1 and Mad2B interacts with the Cdc27 (APC3) subunit of APC/C by recruiting additional Cdc20, thereby increasing APC/C activity to ubiquitylate target proteins. Therefore, Mad2B mediates Cdc20-APC/C ubiquitylation during DDR. Our data identify a previously unknown role for Mad2B-Cdc20-APC/C in DDR.
 XML Download
XML Download