

 PDF
PDF Citation
Citation Print
Print



Introduction
Lipid emulsion (LE) has primarily been used for parenteral nutrition and as a solvent for propofol and etomidate, but is now also used for the treatment of cardiovascular collapse caused by systemic toxicity of local anesthetics [1]. In addition, it has been reported that LE used as an adjuvant drug can treat intractable cardiovascular collapse induced by a toxic dose of non-local anesthetic drugs with high lipid solubility that include calcium channel blocker, beta-blocker, antidepressant, and antipsychotic drugs [2]. Chloroquine (CQ) and hydroxychloroquine are used to treat malaria, systemic lupus erythematosus, and rheumatoid arthritis, and they produce a variety of side-effects that include hypotension, ventricular arrhythmia, QT prolongation, QRS widening, and shock [3–5]. The toxic side-effects of these drugs are due to the blockade of cardiac sodium and potassium channels [3–5]. Hypotension and cardiac depression induced by CQ and hydroxychloroquine toxicity are treated with the following supportive measures: fluid resuscitation, vasopressors (epinephrine, phenylephrine, and norepinephrine), sodium bicarbonate, 3% sodium chloride, dextrose, and activated charcoal [4]. However, in some cases of CQ and hydroxychloroquine toxicity that were unresponsive to supportive treatments, LE was reported to be effective in treating CQ- and hydroxychloroquine-induced toxicity [6–9]. CQ and hydroxychloroquine exhibit the following pharmacological properties that are similar to those of the local anesthetic bupivacaine: First, bupivacaine (log P = 3.41) and CQ (log P = 4.63) are highly lipid-soluble that may contribute to LE-mediated absorption of highly lipid-soluble drugs [1]. Second, CQ and hydroxychloroquine inhibit cardiac sodium and potassium channels, leading to QRS widening, QT prolongation, and cardiac depression that are similar to the symptoms of cardiac toxicity due to bupivacaine toxicity [10,11]. Third, in a rat model of pressure overload hypertrophy, Chaanine et al. [12] showed that CQ-induced cardiac toxicity was mediated by impaired mitochondrial antioxidant function and enhanced oxidative stress. Furthermore, CQ-induced ototoxicity is mediated by reactive oxygen species (ROS) production in glial cells of the ear [13]. Similar to previous reports that demonstrated that CQ toxicity is mediated by oxidative stress and ROS [12,13], bupivacaine-induced cardiac toxicity is also mediated by ROS production, and is inhibited by Intralipid-induced reduction of ROS levels [14,15]. However, the effect of LE on CQ-induced cardiac toxicity remains unknown. Recently, CQ and hydroxychloroquine were reported to attenuate severe acute respiratory syndrome coronavirus 2 in in vitro experiments [16–19]. However, a meta-analysis of further clinical studies reported that treatment with CQ and hydroxychloroquine that were used as alternative drugs to treat patients with coronavirus disease 2019 resulted in cardiac toxicity, including ventricular tachycardia, QT prolongation, torsade de pointes, and cardiac arrest due to the inhibition of sodium and potassium channels in the heart [20,21]. Since bupivacaine and CQ share some pharmacological properties, we tested the biological hypothesis that LE inhibits the cardiac toxicity caused by CQ via inhibition of ROS production or LE-mediated sequestration of highly lipid-soluble CQ [4,10,12–15]. The aim of this in vitro study was to examine the effect of Lipofundin MCT/LCT, an LE, on the cardiac toxicity evoked by CQ in H9c2 rat cardiomyoblasts and clarify the associated cellular mechanism.
Materials and Methods
Cell culture
The American Type Culture Collection supplied the H9c2 cardiomyoblasts (#CRL-1446, USA). H9c2 cells were maintained in high-glucose Dulbecco’s modified Eagle’s medium (DMEM) (#11995, Gibco, Life Technologies, USA) with 10% fetal bovine serum (#16000, Gibco) and 1% penicillin/streptomycin (#15140122, Gibco) at 37°C in a 5% CO2 environment, as reported previously [22]. Prior to drug treatment, the cells were preincubated for 16 h in DMEM without serum.
Cell viability assay
The Cell Counting Kit 8 (CCK-8) (#CK04-13, Dojindo Molecular Technologies, Japan) was used to determine cell viability in accordance with the manufacturer’s procedure, as reported previously [22]. Briefly, H9c2 cells were plated in 24-well dishes at 2 × 104 cells per well and treated for 24 h with various doses of CQ (10-6, 10-5, 10-4, and 10-3 M). To investigate the effect of LE (Lipofundin MCT/LCT) or Intralipid on the decreased cell viability caused by toxic levels of CQ, cells were treated with CQ (10-4 M) alone for 24 h; LE or Intralipid (0.1, 0.2, 0.5, 0.75, and 1%) for 1 h, followed by CQ (10-4 M) for 24 h; or LE or Intralipid (0.1, 0.2, 0.5, 0.75, and 1%) alone for 25 h [23]. In addition, to examine how the ROS scavenger N-acetyl-L-cysteine (NAC) and mitochondrial ROS scavenger mitotempo affect the reduced cell viability produced by toxic amounts of CQ, cells were treated with CQ (10-4 M) alone for 24 h; LE (0.75%) or ROS scavenger (10-4 M NAC or 10-5 M mitotempo) for 1 h, followed by CQ (10-4 M) for 24 h; and LE (0.75%) or ROS scavenger (10-4 M NAC or 10-5 M mitotempo) for 25 h. After the treatment, the cells were incubated with 500 μl of DMEM including 10% CCK-8 solution for 3 h under 5% CO2 at 37℃. The absorbance at 405 nm was determined using a VersaMax® microplate reader (Molecular Devices, USA).
Wound healing assay
A wound healing assay was performed to observe the effects of LE and ROS scavenger on cell migration after CQ treatment of H9c2 cells, as described previously [24]. H9c2 cells were seeded in 100 mm culture dishes (at a density of 1 × 106 cells) and incubated in a CO2 (5% at 37°C) incubator. Cells were grown to 90%–100% confluency. After scratching the cells with a pipette tip (200 μl yellow tip) and washing them with phosphate-buffered saline (PBS), culture medium was added to the culture dishes, and cell images were obtained using a microscope (Nikon Eclipse Ti2; Nikon Co., Japan). Cells were then treated with CQ (10-4 M) alone for 18 h; LE (Lipofundin MCT/LCT, 0.75%) or ROS scavenger (10-4 M NAC or 10-5 M mitotempo) for 1 h, followed by CQ (10-4 M) for 18 h; and LE (0.75%) or ROS scavenger (10-4 M NAC or 10-5 M mitotempo) alone for 19 h. A light microscope fitted with camera (Nikon Eclipse Ti2) was then used to capture images of the cells.
Western blot analysis
Analysis of cleaved caspase-3, Bax, and cleaved caspase-8 protein levels in H9c2 cells was performed using western blotting, as reported previously [22]. For the evaluation of cleaved caspase-3 expression, cells were treated with CQ (10-4 M) alone for 12 h; LE (Lipofundin MCT/LCT, 0.75%) or ROS scavenger (10-4 M NAC or 10-5 M mitotempo) for 1 h, followed by CQ (10-4 M) for 12 h; and LE (0.75%) or ROS scavenger (10-4 M NAC or 10-5 M mitotempo) alone for 13 h. For the evaluation of Bax expression, cells were treated with CQ (10-4 M) for 4 h; LE (0.75%) for 1 h, followed by CQ (10-4 M) for 4 h; or LE (0.75%) alone for 5 h. For the evaluation of cleaved caspase-8 expression, cells were treated with CQ (10-4 M) for 2 h; LE (0.75%) for 1 h, followed by CQ (10-4 M) for 2 h; or LE (0.75%) alone for 3 h. After treatment, the cells were harvested in radio-immunoprecipitation assay lysis buffer (#9806, Cell Signaling Technology, USA) supplemented with protease and phosphatase inhibitor cocktails (#78440, Thermo Fisher Scientific, USA). Protein amounts were quantified using a bicinchoninic acid protein assay reagent kit (#23227, Thermo Fisher Scientific, USA). After 10 min of heating at 100°C, the protein samples were fractionated via 10%–15% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and electrotransferred to polyvinylidene difluoride membranes (#IPVH00010, Millipore, USA). After transfer, membranes were blocked in 5% skim milk or 5% bovine serum albumin in Tris-buffered saline containing 0.5% Tween-20 (TBST) for 1 h at room temperature (22–27℃), followed by incubation with primary antibodies (anti-cleaved caspase-3 [1 : 1000], anti-cleaved caspase-8 [1 : 1000], anti-Bax [1 : 1000], and anti-β-actin [1 : 10000] antibody) for 16 h at 4℃. The membranes were rinsed with TBST, and incubated with horseradish peroxidase-conjugated goat anti-rabbit IgG [1 : 5000] (111-035-003, Jackson ImmunoResearch, USA) or anti-mouse IgG [1 : 5000] (ADI-SAB-100-J, Enzo LifeScience, USA) secondary antibody for 1 h at room temperature (22–27℃). The proteins were visualized using the WesternbrightTM ECL western blotting Detection Kit (#K-12045-D50, Advansta, USA) and ECL images were obtained using a ChemiDocTM Touch Imaging System (Bio-Rad Laboratories Inc., USA). The density of the bands was assessed using ImageJ software (version 1.45s; National Institutes of Health, USA).
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay
Apoptotic cells were identified by TUNEL assay. A TUNEL kit (#11684795910, Roche Applied Science, USA) was used following the manufacturer’s directions as previously reported [22]. H9c2 cells were seeded on coverslips in six-well dishes and treated with CQ (10-4 M) alone for 12 h; LE (Lipofundin MCT/LCT, 0.75%) for 1 h, followed by CQ (10-4 M) for 12 h; or LE (0.75%) alone for 13 h. Cells were then counterstained with 4’,6-diamidino-2-phenylindole (DAPI). Fluorescence images were captured on a fluorescence microscope (Nikon Eclipse Ti2). The number of TUNEL-positive cells was determined as a percentage of DAPI-stained cells in each field of view.
Annexin V-fluorescein isothiocyanate/propidium iodide (Annexin V-FITC/PI) staining
Annexin V-FITC/PI staining was used to assess early/late apoptosis using flow cytometry with a FITC Annexin V Apoptosis Detection Kit (#V13242, Invitrogen-Life Technologies, USA) according to the manufacturer’s protocol, as described previously [22]. Briefly, cells were inoculated in six-well dishes at 1 × 105 cells per well and subsequently treated with CQ (10-4 M) alone for 24 h; LE (Lipofundin MCT/LCT, 0.75%) for 1 h, followed by CQ (10-4 M) for 24 h; or LE (0.75%) alone for 25 h. The cells were collected after treatment, rinsed two times in PBS, and resuspended with 1 × binding buffer. All cells were incubated with annexin V-FITC and a PI staining solution (100 μg/ml) in the dark at room temperature (22–27℃) for 15 min. Binding buffer (1 ×) was added to each tube, and cells were examined with a BD LSRFortessa X-20 flow cytometer (BD Biosciences, USA) to determine the proportion of apoptotic cells in the cell population. The BD FACSDiva™ software (version 6.0; BD Biosciences, USA) was used to analyze the data.
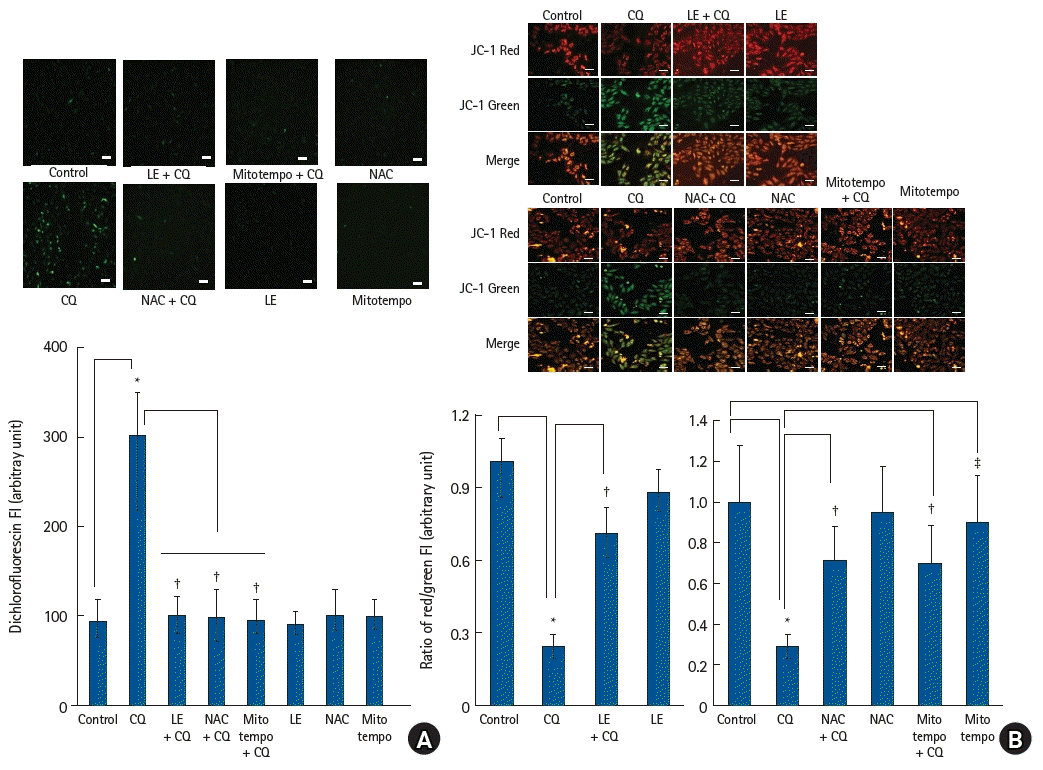
Measurement of intracellular levels of ROS
Intracellular ROS production during CQ treatment was analyzed using 2′, 7′-dichlorofluorescin diacetate (H2DCFDA), as described previously [15]. The cells were seeded in six-well dishes (at a density of 1 × 105 cells per well) containing cover glasses and incubated in a CO2 incubator overnight. The cells were treated with CQ (10-4 M) alone for 1 h; LE (Lipofundin MCT/LCT, 0.75%) or ROS scavenger (10-4 M NAC or 10-5 M mitotempo) for 1 h, followed by CQ (10-4 M) for 1 h; and LE (0.75%) or ROS scavenger (10-4 M NAC or 10-5 M mitotempo) alone for 2 h. Cells were treated with 10 µM H2DCFDA for 30 min at 37°C before being washed with PBS. The mounting solution (#S3023, DAKO fluorescent mounting medium, Dako North America, Inc., USA) was added drop-wise, and the cell-bound cover glass was placed on the slide. Then, images of the cells were obtained using a fluorescence microscope (Nikon Eclipse Ti2) and ROS generation was analyzed.
Mitochondrial membrane potential measurement
The mitochondrial membrane potential was measured using the JC-1 mitochondrial membrane potential detection kit (#30001, Biotium Inc., USA), as previously described [15]. Cells were seeded in six-well dishes containing cover glasses at a density of 1 × 105 cells per well. After incubation, the cells were treated with CQ (10-4 M) alone for 6 h; LE (Lipofundin MCT/LCT, 0.75%) or ROS scavenger (10-4 M NAC or 10-5 M mitotempo) for 1 h, followed by CQ (10-4 M) for 6 h; or LE (0.75%) or ROS scavenger (10-4 M NAC or 10-5 M mitotempo) alone for 7 h. The cells were washed with PBS and then, 1 × JC-1 dye reagent working solution was added to the cells for 15 min, and the cells were washed with PBS. The mitochondrial membrane potential was analyzed using a fluorescence microscope (Nikon Eclipse Ti2) and calculated as the fluorescence intensity of the JC-1 red/green ratio and normalized to the control.
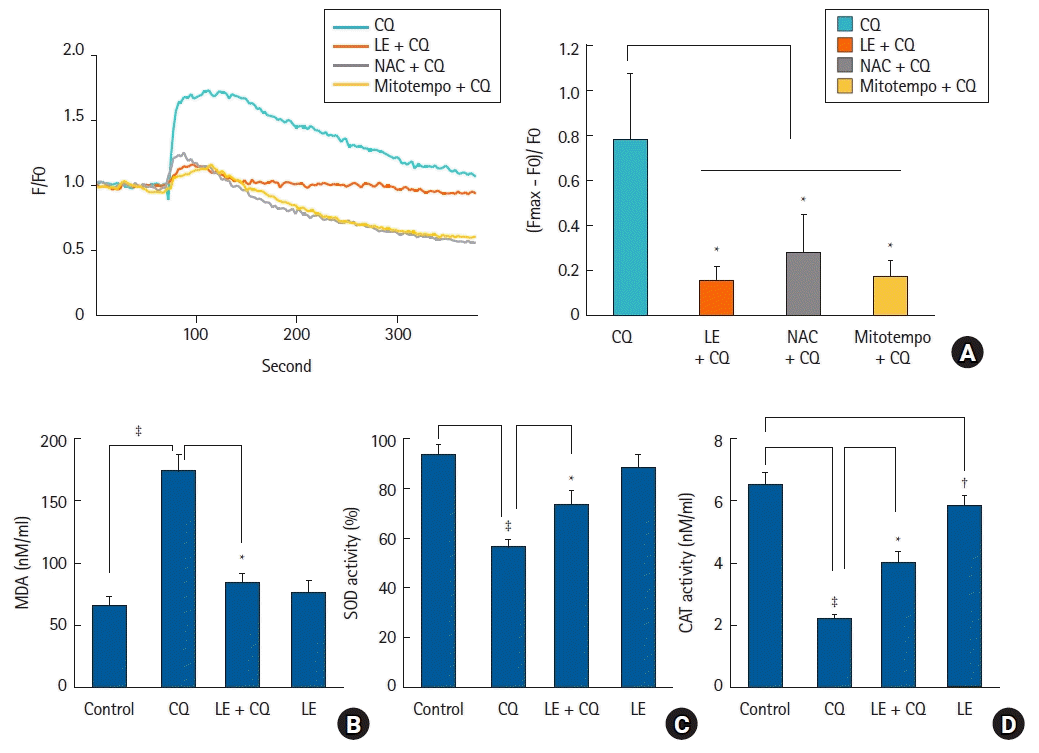
Measurement of intracellular calcium concentration
The change in intracellular calcium levels ([Ca2+]i) was assessed using a confocal laser microscope (IX70 Fluoview, Olympus, Tokyo, Japan), as described previously [15]. H9c2 cells were cultured in a confocal cell culture dish (SPL, Korea) and incubated with 2.5 μM Fluo-4 AM (#F14217, Invitrogen-Life Technologies) in Hank’s Balanced Salt Solution (HBSS, #14175, Gibco) for 30 min. Subsequently, cells were washed twice with PBS and after replacing the HBSS, the cells were treated with CQ (10-4 M) alone and LE (Lipofundin MCT/LCT, 0.75%) or ROS scavenger (10-4 M NAC or 10-5 M mitotempo), followed by CQ (10-4 M). Ca2+ levels were monitored every 2.5 s at excitation and emission wavelengths of 485 and 520 nm, respectively. Ca2+ levels were analyzed in scanned images and calculated as drug-treated fluorescence intensity (F) divided by baseline fluorescence intensity (F0) before drug treatment. The net change in calcium ions was indicated as (Fmax-F0) / F0, where Fmax represents the maximum calcium level corresponding to the fluorescence intensity after CQ (10-4 M) treatment in the presence or absence of LE or ROS scavenger pretreatment. Changes in Ca2+ levels were measured for 7 min.
Measurement of malondialdehyde (MDA), superoxide dismutase (SOD), and catalase (CAT) activity
The levels of MDA (#ab118970), SOD (#ab65354), and CAT (#ab83464) were measured with MDA, SOD, and CAT assay kits (Abcam, USA), respectively, according to the manufacturers’ protocols, as described previously [25]. To measure SOD, MDA, and CAT activity, H9c2 cells were cultured in 100 mm culture dishes at a density of 2 × 106, 1 × 106, and 1 × 106 cells, respectively. Cells were treated with CQ (10-4 M) alone for 18 h, LE (Lipofundin MCT/LCT, 0.75%) for 1 h followed by CQ (10-4 M) for 18 h, or LE (0.75%) alone for 19 h. The samples collected for MDA activity measurement were incubated with MDA lysis buffer, homogenized, and centrifuged to collect the supernatant. Samples were incubated with thiobarbituric acid reagent at 95℃ for 60 min. After allowing the samples to cool to room temperature (22–27℃) on ice, the analysis was performed with a VersaMax® microplate reader. The MDA concentration was calculated using the formula provided with the assay kit. The harvested cells for SOD activity measurement were lysed in lysis buffer and centrifuged to collect the supernatant. SOD activity was measured using water-soluble tetrazolium salts working solution, dilution buffer, and enzyme working solution. SOD activity was measured at 450 nm using a VersaMax® microplate reader. The SOD level was calculated using the formula described in the assay kit. Samples for CAT activity measurement were lysed in CAT assay buffer and the supernatant was collected. For the CAT reaction, H2O2 was added to the samples and incubated at 25°C, followed by the addition of stop solution. The prepared Developer Mix was mixed with the samples and incubated at 25℃. CAT activity was measured at 570 nm using a VersaMax® microplate reader, and calculated using the formula provided with the assay kit.
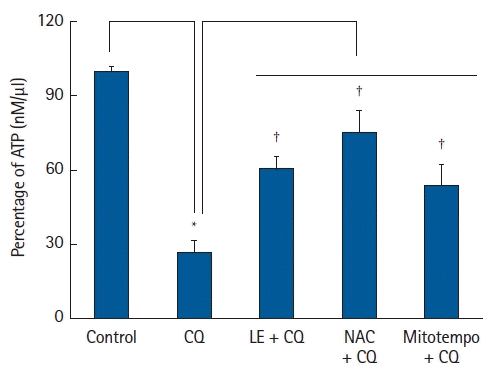
Adenosine triphosphate (ATP) measurement
Intracellular ATP levels in CQ-treated H9c2 cells were measured using an ATP detection kit (#ab83355, Abcam) according to protocol of the manufacturer, as described previously [26]. H9c2 cells were seeded and incubated in a serum-free medium in a 37°C CO2 incubator. Cells were treated with CQ (10-4 M) alone for 18 h and LE (Lipofundin MCT/LCT, 0.75%) or ROS scavenger (10-4 M NAC or 10-5 M mitotempo) for 1 h, followed by CQ (10-4 M) for 18 h. Cells were lysed with ATP assay buffer, and centrifuged (13,000 × g) at 4°C for 5 min to collect the supernatant and transferred to a new tube. The prepared ATP reaction mixture was added to the samples and standards, then, mixed and incubated at room temperature (22–27℃) for 30 min in the dark. ATP levels were measured using a VersaMax® microplate reader at 570 nm, and a standard curve was created with authentic ATP for quantitative analysis.
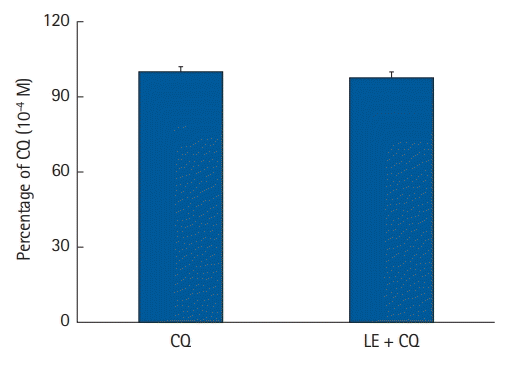
Effect of LE on CQ concentration in the distilled water
CQ (10-4 M) was emulsified with LE (Lipofundin MCT/LCT, 0.75%) in distilled water, as described earlier [15]. The sample emulsion was centrifuged at 18,500 × g for 30 min to estimate the release of CQ from the samples. The concentration of CQ in the aqueous phase was analyzed using ultra-performance liquid chromatography-quadrupole time-of-flight mass spectrometry (UPLC-Q-TOF MS; Waters, USA). The aqueous layer was injected into an Acquity UPLC BEH C18 column (100 × 2.1 mm, 1.7 m; Waters) equilibrated with water/acetonitrile (99 : 1), including 0.1% formic acid (FA), and dissolved in a linear gradient (1%–100%) of acetonitrile, including 0.1% FA, for 5 min at a flow rate of 0.35 ml/min. Q-TOF MS in positive electrospray ionization mode was used to evaluate the eluted CQ. The optimal MS operating conditions were set as follows: The voltages of capillary and sampling cones were set to 3 V and 30 kV, respectively; the source and desolvation temperatures were 400 and 100°C, respectively; and the desolvation flow was 800 L/h. The eluted CQ was detected by multiple reaction monitoring mode with precursor and product ions of m/z 409.14 and 294.08, respectively. To assure the reproducibility and accuracy of all analyses, a lock spray was performed with leucine-enkephalin ([M + H] = 556.2771) used as the lock mass. All mass results were gathered and evaluated using the UIFI 1.8.2 (Waters).
Materials
The purest chemical substances are commercially available. Lipofundin MCT/LCT 20% was donated by B. Braun Melsungen AG (Germany). Intralipid 20% was obtained from Fresenius Kabi AB (Sweden). CQ (#PHR1258), anti-β-actin antibody (#A5441), NAC (#A9165), mitotempo (#SML0737), DAPI (#D9542), and H2DCFDA (#287810) were purchased from Sigma-Aldrich (USA). The anti-cleaved caspase-8 antibody was purchased from Novus Biologicals (#NB100-56116, USA). Anti-cleaved caspase-3 (#9661) and anti-Bax (#2772) antibodies were obtained from Cell Signaling Technology. All drugs were dissolved in distilled water.
Statistical analysis
Normality tests were performed using a Kolmogorov-Smirnov or Shapiro-Wilk test. The primary end points were the effects of LE, Intralipid, and ROS inhibitors on cell viability and migration, TUNEL-positive cells, apoptosis, cleaved caspase-3, cleaved caspase-8, and Bax expression that are induced by CQ. The effects of LE, Intralipid, CQ, and ROS inhibitors, alone or in combination, on cell viability and migration, TUNEL-positive cells, apoptosis, cleaved caspase-3, cleaved caspase-8, Bax expression, mitochondrial membrane potential, intracellular calcium level, MDA, SOD and CAT activity, and ATP were analyzed using a one-way analysis of variance, followed by Bonferroni’s test (Prism, Version 5.0; GraphPad Software, USA). The effects of LE and CQ, alone or in combination, on mitochondrial membrane potential were analyzed using the Kruskal-Wallis test, followed by Dunn’s multiple comparison test. The Kruskal-Wallis test, followed by Dunn’s multiple comparison test was used to analyze the effects of LE, CQ, and ROS inhibitors, alone or in combination, on ROS production. An unpaired Student’s t-test was used to analyze the effect of LE on the CQ concentration. The number of independent experiments was determined using the resource equation method and based on previous similar experiments [25,27]. Results with P < 0.05 were considered statistically significant.
Results
Effects of LE and ROS scavenger on the decreased cell viability and migration induced by a toxic dose of CQ
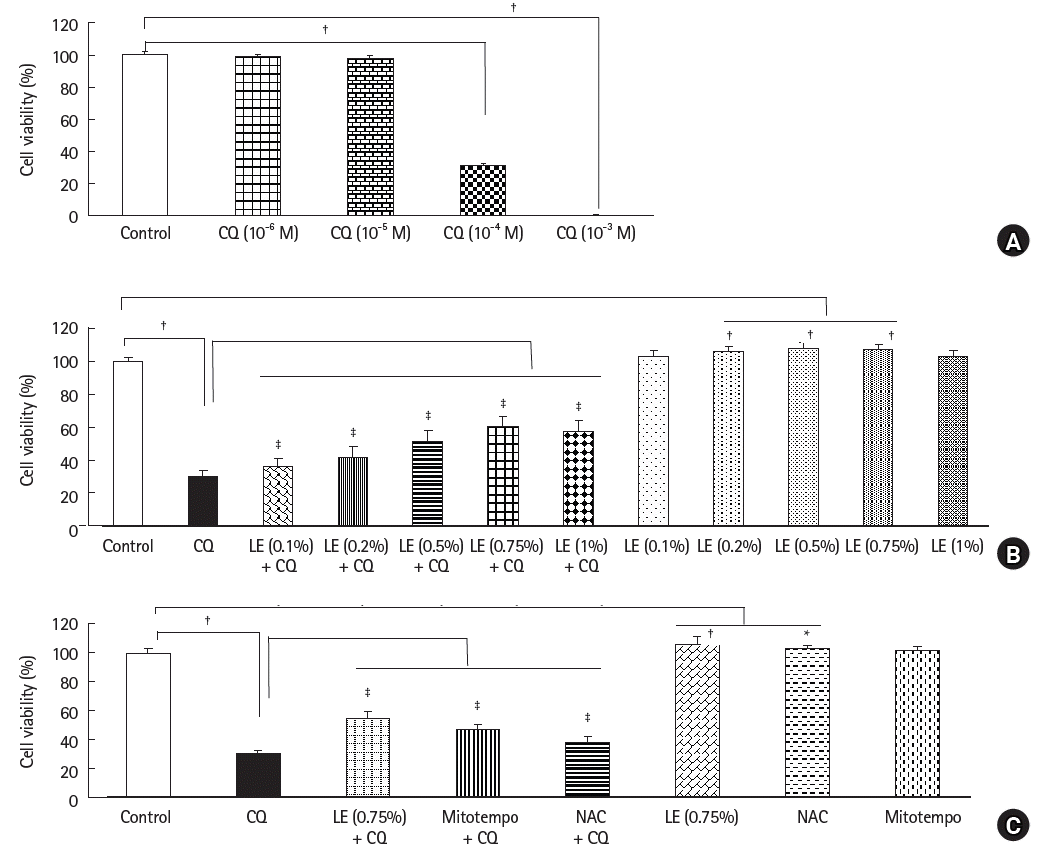
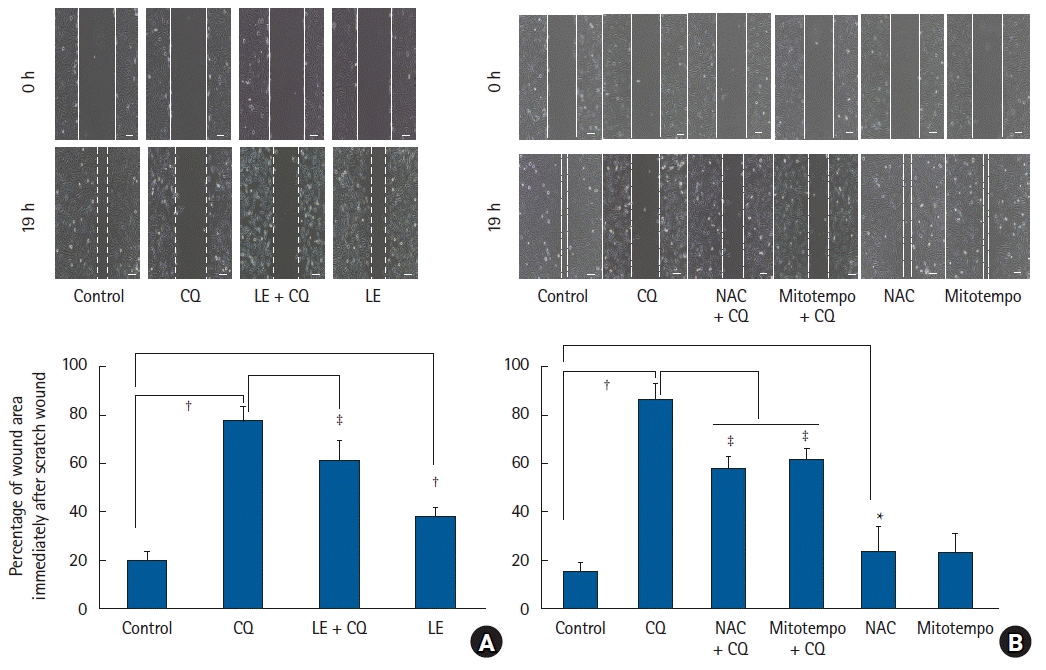
Treatment with CQ (10-4 and 10-3 M) reduced cell viability (P < 0.001 versus the control, 95% CI: 66.78 to 70.86 and 97.61 to 101.7 at 10-4 and 10-3 M, respectively, Fig. 1A). Lipofundin MCT/LCT (0.1 to 1%) that is made up of 50% long-chain triglycerides and 50% medium-chain triglycerides reversed the decrease in cell viability induced by CQ (10-4 M) (0.1 to 1%: P < 0.001 versus CQ alone, 95% CI: −10.0 to −2.1, −15.1 to −7.2, -25.0 to −17.0, −34.2 to −26.8, and −30.8 to −23.0 at 0.1%, 0.2%, 0.5%, 0.75%, and 1%, respectively, Fig. 1B). In contrast, Intralipid (0.2% to 1%) that contains only 100% long-chain triglyceride only slightly reversed the CQ (10-4 M)-induced reduction in cell viability (0.2%: P = 0.012 versus CQ alone, 0.5% to 1%: P < 0.001 versus control, 95% CI: −8.2 to −0.6, −11.3 to −3.7, −15.1 to −7.5, and −17.5 to −9.9 at 0.2, 0.5, 0.75, and 1%, respectively, Supplementary Fig. 1). Lipofundin MCT/LCT (0.2%, 0.5%, and 0.75%) alone slightly improved cell viability (0.2%, 0.5%, and 0.75%: P < 0.001 versus control, 95% CI: −10.2 to −2.3, −12.2 to −4.3, and −11.4 to −3.5 at 0.2%, 0.5%, and 0.75%, respectively, Fig. 1B), but Intralipid (0.1%, 0.2%, 0.75%, and 1%) alone reduced cell viability (0.75%: P = 0.006 versus control, 0.1%, 0.2%, and 1%: P < 0.001 versus control, 95% CI: 3.7 to 11.4, 2.3 to 10.0, 1.0 to 8.6, and 2.6 to 10.2 at 0.1%, 0.2%, 0.75%, and 1%, respectively, Supplementary Fig. 1). The ROS scavenger NAC (10-4 M) and mitochondrial ROS scavenger mitotempo (10-5 M) reversed the reduction in cell viability evoked by CQ (10-4 M) (P < 0.001 versus CQ alone; 95% CI: −19.7 to −14.0 and −10.9 to −5.2 at mitotempo and NAC, respectively, Fig. 1C). In addition, CQ treatment (10-4 M) reduced cell migration (P < 0.001 versus the control; 95% CI: −64.7 to −50.0, Fig. 2A). However, pretreatment with LE (0.75%) increased cell migration compared with CQ alone (P < 0.001; 95% CI: 9.2 to 23.9, Fig. 2A). Additionally, pretreatment with the ROS scavenger NAC (10-4 M) or mitotempo (10-5 M) increased cell migration compared to CQ alone (P < 0.001; 95% CI: 20.3 to 36.8 and 16.4 to 32.9 at NAC and mitotempo, respectively, Fig. 2B).
Effects of LE and ROS scavenger on apoptosis induced by a toxic dose of CQ
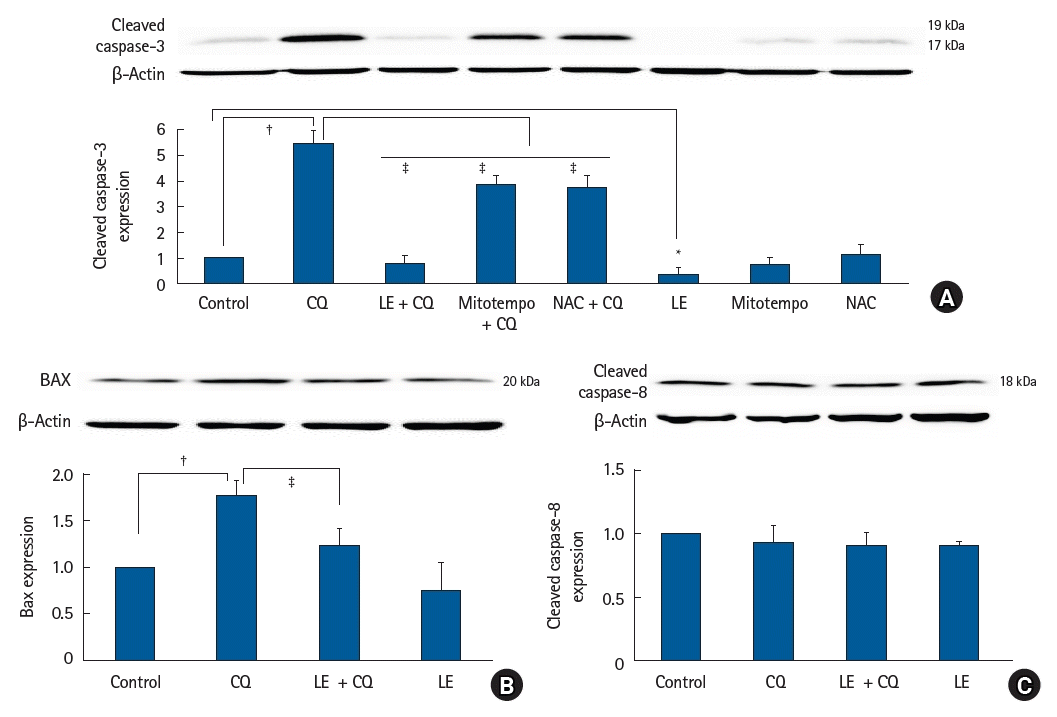
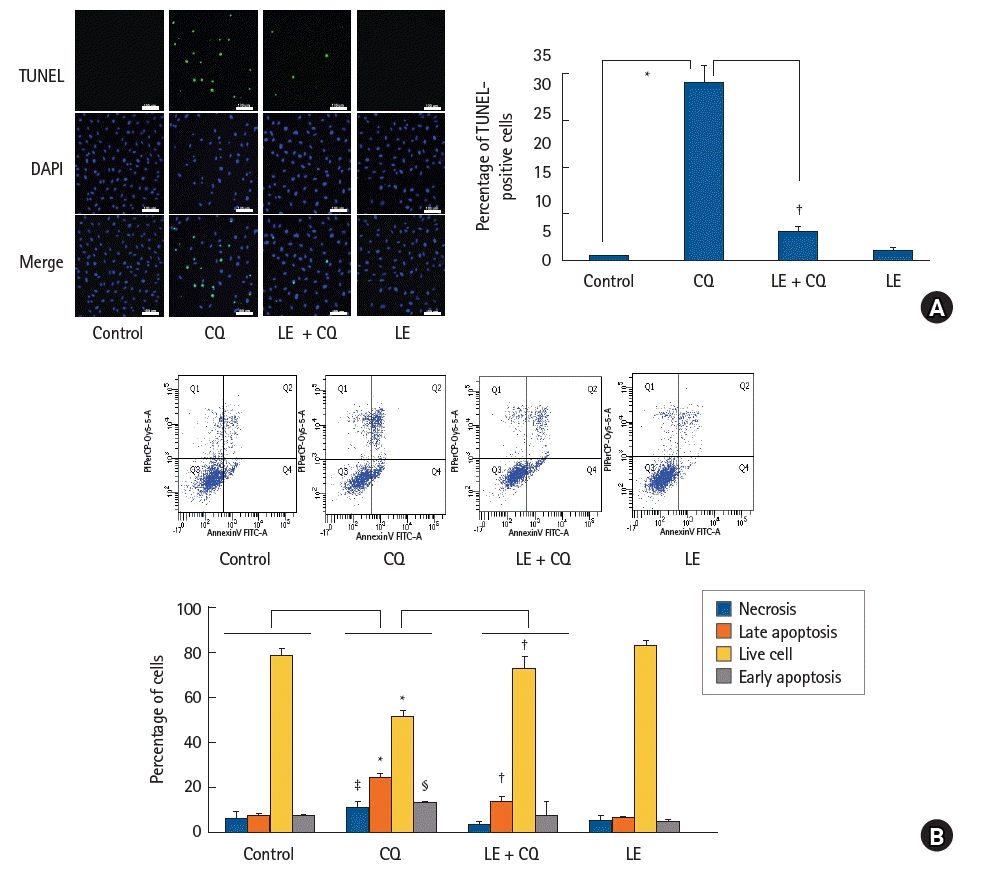
The cleavage of caspase-3 (a signaling molecule downstream of the common pathway associated with both intrinsic and extrinsic apoptotic cell death) is a well-known marker of the apoptotic cell death process [28]. Treatment with CQ (10-4 M) induced cleaved caspase-3 expression (P < 0.001 versus control; 95% CI: −4.94 to −3.96, Fig. 3A), whereas LE (0.75%), ROS scavenger NAC (10-4 M), and mitotempo (10-5 M) treatment inhibited the CQ (10-4 M)-induced cleaved caspase-3 expression (P < 0.001 versus CQ alone; 95% CI: 4.14 to 5.15, 1.09 to 2.07, and 1.2 to 2.17 at LE, mitotempo, and NAC, respectively, Fig. 3A), leading to inhibition of apoptotic cell death. CQ (10-4 M) produced an increase in the expression of the intrinsic proapoptotic protein Bax (P < 0.001 versus control; 95% CI: −1.043 to −0.4667, Fig 3B) that is associated with mitochondrial stress [28]. LE treatment (0.75%) reduced the increase in Bax expression induced by CQ (10-4 M) (P < 0.001 versus CQ alone; 95% CI: 0.23 to 0.81, Fig. 3B), suggesting that it reduces mitochondrial stress. However, CQ treatment (10-4 M) did not significantly affect the expression of the extrinsic proapoptotic protein cleaved caspase-8 that is associated with apoptotic cell death activated via cell death receptors [28], and LE did not significantly affect the expression of cleaved caspase-8 induced by CQ (10-4 M) (Fig. 3C). CQ (10-4 M) treatment induced TUNEL-positive cells (P < 0.001 versus the control; 95% CI: −32.6 to −27.7, Fig. 4A), suggesting the presence of DNA fragmentation due to apoptosis, whereas LE (0.75%) treatment inhibited the CQ (10-4 M)-induced increase in TUNEL-positive cells (P < 0.001 versus CQ alone; 95% CI: 23.5 to 28.3, Fig. 4A), suggesting inhibition of apoptosis. Furthermore, CQ (10-4 M) treatment increased both early and late stages of apoptotic cell death (early apoptosis: P = 0.005 versus control, late apoptosis: P < 0.001 versus control; 95% CI: −9.53 to −1.97 and −19.3 to −14 at early and late apoptosis, respectively, Fig. 4B). CQ (10-4 M) increased necrosis (P = 0.034, 95% CI: −8.7 to −0.2, Fig. 4B), however, LE (0.75%) treatment inhibited the late stage of apoptotic cell death evoked by CQ (10-4 M) (P < 0.001 versus CQ alone; 95% CI: 8.1 to 13.4, Fig. 4B).
Effects of LE and ROS scavengers on physiological changes induced by toxic dose of CQ
CQ treatment (10-4 M) increased ROS levels (P < 0.001 versus control, Fig. 5A), whereas pretreatment with LE (0.75%), the ROS scavenger NAC (10-4 M), and mitotempo (10-5 M) inhibited the ROS production evoked by CQ (10-4 M) (P < 0.001 versus CQ alone; Fig. 5A). CQ treatment (10-4 M) reduced the mitochondrial membrane potential (P < 0.001 versus control; Fig. 5B), suggesting that it promotes mitochondrial dysfunction. However, treatment with LE (0.75%), NAC (10-4 M), and mitotempo (10-5 M) reversed the reduction in mitochondrial membrane potential evoked by CQ (10-4 M) (P < 0.001 versus CQ alone; 95% CI: −0.608 to −0.384 and −0.592 to −0.368 at NAC and mitotempo, respectively, Fig 5B), indicating the attenuation of mitochondrial dysfunction. CQ treatment (10-4 M) increased the intracellular calcium levels (Fig. 6A), suggesting that calcium regulation is impaired by this drug. However, treatment with LE (0.75%), the ROS scavenger NAC (10-4 M), and mitotempo (10-5 M) reduced the calcium levels increased by CQ (10-4 M) (P < 0.001 vs. CQ alone; 95% CI: 0.50 to 1.34, 0.47 to 1.30, and 0.81 to 1.64 at LE, NAC, and mitotempo, respectively, Fig. 6A), suggesting that they reduce the mitochondrial ROS-induced increase in calcium. CQ treatment (10-4 M) increased the levels of MDA, an indicator of oxidative stress (P < 0.001 versus control; 95% CI: −120 to −71, Fig. 6B), whereas LE treatment (0.75%) inhibited the increased MDA activity evoked by CQ (10-4 M) (P < 0.001 versus CQ alone; 95% CI: 55 to 104, Fig. 6B), suggesting an inhibition of increased oxidative stress. CQ treatment (10-4 M) reduced the activities of SOD and CAT (SOD and CAT: P < 0.001 versus control; 95% CI: 29.5 to 41.8 and 3.7 to 4.7 for SOD and CAT, respectively, Figs. 6C and D) that are antioxidant enzymes, suggesting a relative increase in oxidative stress. However, LE (0.75%) reversed the reduction in the activities of SOD and CAT induced by CQ (10-4 M) (P < 0.001 versus CQ alone; 95% CI: −21.9 to −9.5 and −2.3 to −1.3 for SOD and CAT, respectively, Figs. 6C and D), suggesting that it promotes relatively decreased oxidative stress.
Effects of LE and ROS scavengers on ATP levels decreased by toxic dose of CQ
Additionally, CQ treatment (10-4 M) reduced ATP levels (P < 0.001 versus the control; 95% CI: 61.16 to 84.39, Fig. 7). However, treatment with LE (0.75%), the ROS scavenger mitotempo (10-5 M), and NAC (10-4 M) reversed the reduction in ATP levels evoked by CQ (10-4 M) (P < 0.001 versus CQ alone; 95% CI: −44.93 to −22.70, −59.71 to −37.47, and −37.87 to −15.63 for LE, NAC, and mitotempo, respectively, Fig. 7).
Effect of LE on CQ concentration
LE (0.75%) did not significantly alter CQ concentration (10-4 M) in distilled water (P = 0.0572, Fig. 8), suggesting that LE does not absorb CQ.
Discussion
This study demonstrates that LE inhibits the cardiac toxicity caused by CQ toxicity, and that this is due to the direct inhibition of ROS production. The major results of this study are as follows: 1) treatment with LE and the ROS scavengers NAC and mitotempo reversed the reduction in cell viability and migration and increase in cleaved caspase-3 expression evoked by CQ; 2) LE treatment inhibited Bax expression and the late apoptosis induced by CQ; 3) treatment with LE and ROS scavengers reversed ROS production, increased calcium levels, and reduced ATP levels and mitochondrial membrane depolarization caused by CQ.
Intralipid reverses the reduction in cell viability induced by verapamil, amlodipine, and bupivacaine that are highly lipid soluble (log P > 2) [14,15,22,29,30]. In addition, NAC attenuates the bupivacaine-induced reduction in cell viability [15]. In line with earlier reports, LE, NAC, and the mitochondrial ROS scavenger mitotempo reversed the reduction in cell viability evoked by toxic levels of highly lipid-soluble CQ that suggests that the ROS production induced by CQ contributes to reduced cell viability [14,15,23]. In accordance with the results of the cell viability study, CQ treatment reduced cell migration, whereas LE, NAC, and mitotempo treatment reversed the reduction in cell migration induced by CQ. Intralipid inhibits cleaved caspase-3 expression in rat cardiomyoblasts exposed to a toxic dose of bupivacaine, suggesting that Intralipid inhibits apoptotic cell death [31]. Similar to this report, LE, NAC, and mitotempo inhibited the cleaved caspase-3 expression induced by toxic concentrations of CQ [31]. In addition, LE inhibited the Bax expression induced by toxic concentrations of CQ; however, a toxic dose of CQ did not significantly change cleaved caspase-8 expression. When combined, these results suggest that LE inhibits CQ-evoked apoptosis via the intrinsic mitochondrial apoptotic pathway that is mediated by the inhibition of ROS production. Compared to Lipofundin MCT/LCT, Intralipid displays greater inhibition of the decrease in cell viability induced by a toxic dose of bupivacaine [31]. However, in the current study, Lipofundin MCT/LCT substantially reversed the decrease in cell viability due to a toxic dose of CQ in comparison to Intralipid. The differences in these results may be due to specific physicochemical interactions between the fatty acids (100% long-chain alone or 50% long-chain plus 50% medium-chain) and the offending drugs (bupivacaine or CQ), as well as the different concentrations of Intralipid and Lipofundin MCT/LCT used. The Lipofundin MCT/LCT-mediated increased reversal of reduced cell viability caused by CQ may be associated with medium-chain triglycerides contained in Lipofundin MCT/LCT, compared to Intralipid (that is comprised of 100% long-chain fatty acid alone). In addition, this difference may be ascribed to the differences in cell viability evoked by Lipofundin MCT/LCT or Intralipid alone (increased cell viability evoked by Lipofundin MCT/LCT alone versus decreased cell viability evoked by Intralipid alone; Fig. 1B versus Supplementary Fig. 1). Further studies should examine which type of medium-chain triglyceride present in Lipofundin MCT/LCT is the main contributor to the reversal of reduced cell viability.
Intracellular ROS that are mostly produced in mitochondria induce disturbances in respiratory chain function, leading to further ROS generation, a reduced membrane potential of mitochondria, and reduced ATP production [28]. Eventually, ROS in the mitochondria that are produced endogenously or exogenously induce p53 and c-Jun N-terminal kinase activation, leading to the activation of proapoptotic Bcl-2 proteins (Bad, Bax, Puma, Noxa, and Bak) [28]. In addition, ROS induces cardiolipin oxidation, leading to cytochrome c release and mitochondrial membrane depolarization [28]. Intralipid, NAC, and mitotempo have been shown to reduce ROS production and mitochondrial membrane depolarization induced by a toxic dose of bupivacaine [14,15]. Similar to previous reports involving bupivacaine, LE and the ROS scavengers NAC and mitotempo inhibited the ROS production and mitochondrial membrane depolarization induced by CQ in rat cardiomyoblasts [12–15,25]. Combined with these results, it suggests that the LE-mediated attenuation of mitochondrial membrane depolarization caused by CQ is mediated by a decrease in ROS production. In agreement with the LE-evoked reduction in ROS produced by CQ, LE reversed the CQ-induced increase in the activity of MDA that is produced via lipid peroxidation by free radicals [32]. However, LE reversed the CQ-induced reduction of activities of the antioxidant enzymes SOD and CAT. Taken together, the LE-mediated reduction of ROS produced by CQ mainly contributes to the attenuation of cardiotoxicity induced by a toxic level of CQ. ATP treatment nearly reversed the decreased myocardial contractility caused by bupivacaine [33]. Bupivacaine has been shown to attenuate cardiac mitochondrial ATP synthesis [34,35], and Intralipid contributes to the attenuation of bupivacaine-induced cardiotoxicity by stimulating fatty acid oxidation that is associated with ATP production [36]. Taken together, these previous studies suggest that fatty acid oxidation and subsequently ATP production mediated by Intralipid attenuates bupivacaine-induced cardiotoxicity [33–36]. Similar to these reports regarding bupivacaine-induced cardiotoxicity [33–36], CQ (10-4 M)-evoked ROS production reduces ATP production, whereas LE, NAC, and mitotempo treatment reversed the reduction in ATP production induced by a toxic level of CQ (10-4 M) via the inhibition of ROS production [14,15,33–36]. Further studies are needed to investigate detailed upstream and downstream (for example, mitochondrial cytochrome c release) cellular signaling pathways associated with the CQ-induced ROS production in cardiomyoblasts. Mitochondria are important for maintaining calcium homeostasis [28]. ROS production in the mitochondria disrupts calcium homeostasis in the intracellular compartment, leading to increased intracellular calcium levels [37]. Intralipid was shown to increase calcium retention capacity and subsequently inhibited mitochondrial permeability transition pore opening in bupivacaine-induced cardiotoxicity, suggesting that Intralipid provides better control of calcium overload [36]. Intralipid, NAC, and mitotempo reduce the calcium level increased by toxic dose of bupivacaine [15]. Comparable to these reports [15,36], our results indicated that LE, NAC, and mitotempo inhibited the increase in intracellular calcium levels induced by CQ treatment (10-4 M). When combined with these results, it suggests that LE-mediated inhibition of ROS production by CQ contributes to the inhibition of increased intracellular calcium levels caused by the toxic concentration of CQ.
The widely accepted underlying mechanism of LE resuscitation for drug toxicity caused by drugs with high lipid solubility is an indirect ‘lipid shuttle,’ in which the lipid compartment of LE absorbs lipid-soluble drugs (for example, bupivacaine; log P of bupivacaine = 3.41) from the heart and brain, and then LEs containing lipid-soluble drugs are delivered to the muscle, adipose tissue, and liver for storage and detoxification [38]. In addition, Intralipid (0.25%, 1%, and 2%) has been shown to decrease bupivacaine concentration [39]. As the lipid solubility of CQ (log P = 4.63) is higher than that of bupivacaine (log P = 3.41), high-performance liquid chromatography was carried out to examine whether the lipid compartment of LE absorbed highly lipid-soluble CQ. However, as LE (0.75%) did not significantly reduce CQ (10-4 M) concentration, we surmise that the LE-evoked reduction in ROS generation induced by CQ is due to the direct inhibition of CQ-induced ROS production by LE. The direct underlying mechanisms of LE resuscitation include inotropic effects, reversal of mitochondrial dysfunction, attenuation of ischemic reperfusion injury, and inhibition of nitric oxide release [1]. These observations indicate that the underlying mechanism associated with the LE-evoked reversal of cardiotoxicity induced by CQ toxicity is associated with the reversal of mitochondrial dysfunction via the inhibition of ROS production [1].
This study has some limitations. First, the current study did not examine the effect of LE on the cardiac dysfunction caused by a toxic dose of CQ in an in vivo model. High CQ concentrations of 1 and 3 × 10-5 M have been shown to decrease left ventricular pressure and heart rate, respectively, in rats [40]. Furthermore (and similar to our results in the current study), Blignaut et al. [40] also demonstrated that a high dose of CQ (1 × 10-4 M) decreased the cell viability of isolated ventricular myocytes [40]. However, further investigation will be needed to confirm these findings in vivo. Second, LE was administered prior to treatment with a toxic dose of CQ in this study, whereas LE is clinically administered after the cardiovascular collapse evoked by a toxic dose of CQ. Third, the H9c2 rat cardiomyoblast cell line was used in this experiment, but primary cultured cardiomyocytes are more clinically relevant. Despite these limitations, considering that CQ and hydroxychloroquine have similar pharmacological and toxicological properties, this study suggests that LE treatment may be helpful to treat critical patients with cardiac toxicity due to CQ or hydroxychloroquine toxicity, who need emergency anesthesia and intensive care. Furthermore, LE may be effective in treating the cardiotoxicity observed after CQ and hydroxychloroquine treatment in patients suffering from coronavirus disease 2019 [20,21]. LE was reported to be effective in alleviating cardiovascular depression caused by a toxic dose of highly lipid soluble drugs that inhibit cardiac sodium or potassium channels that include amitriptyline, bupropion, lamotrigine, and flecainide [10,41,42]. Thus, LE may be a valuable adjuvant drug in the treatment of patients with intractable cardiovascular depression caused by toxicity of the afore-mentioned drugs in the operating room or intensive care unit. In addition, further studies are needed to examine the effect of LE alone on the cardiovascular system and elucidate the details of the underlying mechanisms.
In conclusion, LE with fatty acids that contain 50% long-chain and 50% medium-chain inhibited the late apoptosis and cardiotoxicity caused by CQ toxicity that is mediated by the direct reduction of ROS production in the mitochondrial intrinsic apoptotic pathway of rat cardiomyoblasts. LE as an adjuvant drug with supportive treatments may be effective in treating intractable cardiac toxicity induced by CQ.
 XML Download
XML Download