

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Numerous international and national cohort data have shown continued growth in both the incidence and prevalence of chronic kidney disease (CKD) and subsequent end-stage kidney disease (ESKD) [1-3]. An increasing number of studies worldwide has reported on the growing prevalence of CKD; one of these studies pooled the results of 33 population-based representative studies from around the world and reported an age-standardized global prevalence values of CKD stages 1–5 in individuals aged ≥20 years of 10.4% among men and 11.8% among women in 2010 [3,4]. Due in part to the rise in causative factors such as diabetes and obesity, the number of patients affected by CKD has continued to rise, coming to affect an estimated 843.6 million individuals worldwide in 2017 [3,5]. In Korea, the total number of new ESKD patients starting kidney replacement therapy (KRT) was approximately 10,000 in 2011, while the total number of new ESKD patients who started KRT for ESKD was almost 19,000 in 2019 [2], indicating an overwhelmingly increasing trend in ESKD patients. Beyond the socio-economic problems that have arisen for CKD and ESKD burdens, CKD has come to be recognized as one of the leading causes of death worldwide [3]. The Global Burden of Disease report has predicted that CKD will become the fifth-highest cause of years of life lost globally by 2040 [6]. These findings may suggest that previous and current strategies for CKD prevention and management do not have the strength to hold out much longer.
In fact, clinicians have long had insufficient tools at their disposal with which to address CKD. For decades, the struggle has felt like a battle against a Goliath in the disease, where the only ‘stone’ that clinicians have had to throw has been renin-angiotensin blockade, such as through the use of angiotensin-converting enzyme (ACE) inhibitor and angiotensin receptor blocker. Despite this, there have been fewer interventional trials to develop therapeutics to stop the progression of kidney disease than those intended to address almost any other medical subspecialty, and as a result there are relatively few therapies for the treatment of CKD at present [7,8]. Although there have been certain trials for the development of novel therapeutics, attempts to achieve improved renal protection using new targets for CKD have been disappointing [8]. Several potential agents such as protein kinase C (PKC)-β inhibitor, endothelin type A receptor antagonist, pirfenidone, sulodexide, direct renin inhibitor, and nuclear 1 factor (erythroid-derived 2)-related factor 2 activator have all been suggested and introduced as new therapy of CKD but eventually most of them did not fail to cross the threshold of efficacy and safety [9-14]. Since existing therapy falls far short of inducing remission of CKD or even halting progression to ESKD, there has been continued urgency over novel therapeutics to slow the overall increase in CKD and ESKD. Fortunately, new help has emerged on the horizon.
Go to : 
KIDNEY PROTECTION WITH SODIUMGLUCOSE COTRANSPORTER-2 INHIBHITOR
Sodium-glucose cotransporter-2 inhibitor is not just a glucose-lowering agent
The hope for CKD sufferers came from an extremely unexpected source after scientists and researchers identified the excellent benefits of a glucose-lowering agent that has been shown to be effective in delaying and even preventing the progression of kidney disease: the ‘sodium-glucose cotransporter-2 (SGLT2) inhibitor.’ The robust renal and cardiac benefits that are now being observed in this inhibitor were not widely predicted when the first SGLT2 inhibitors were being trialed [15]. Solid evidences, ranging from the BI 10773 (Empagliflozin) Cardiovascular Outcome Event Trial in Type 2 Diabetes Mellitus Patients (EMPA-REG OUTCOME) trial to The Study of Heart and Kidney Protection With Empagliflozin (EMPA-KIDNEY) trial, have indicated that SGLT2 inhibitors play major roles in slowing the progression of kidney disease clearly, consistently, and coherently [16-18]. An updated meta-analysis of 13 major randomized trials of SGLT2 inhibitors has comprehensively shown that SGLT2 inhibitors significantly improve robust kidney endpoints, including a standardized, composite definition of kidney disease progression applied to all trials [15,19]. In this analysis, SGLT2 inhibitors were found to safely reduce the risk of kidney disease progression by 37% (relative risk [RR], 0.63; 95% confidence interval [CI], 0.58 to 0.69), compared to placebo, with similar reductions seen in patients with and without diabetes [19].
Based on these points of advanced evidence, the Kidney Disease: Improving Global Outcomes (KDIGO) clinical practice guideline for diabetes management in CKD had to be updated faster than expected [20,21]. One of the critical parts of the change in the KDIGO 2022 guideline is that SGLT2 inhibitors are not considered solely a glucose-lowering agent, as practitioners are now instructed to treat them as an essential component of CKD care beyond glycemic effects [21]. Another change includes that SGLT2 inhibitors are now recommended to be administered to type 2 diabetes patients with an estimated glomerular filtration rate (eGFR) down to ≥20 mL/min/1.73 m2 from a previous guideline of ≥30 mL/min/1.73 m2 [21]. These recommendations have since been adopted in the Korean Society of Nephrology 2023 practical recommendations for the management of diabetic kidney disease [22].
The precise mechanisms by which SGLT2 inhibitors provide kidney benefits remains a subject of debate
Kidney benefits by SGLT2 inhibitors appear independent of glucose-lowering, and the reduction of intraglomerular pressure, along with single-nephron hyperfiltration, have been suggested to be the theoretical mechanism for their renal protection [21,23]. Is this explanation sufficiently convincing? If so, why do other diuretics not have similar renoprotective effects? A complete interpretation of the mechanism appears to still be elusive. In retrospect, when such inhibitors were first introduced, some nephrologists speculated that—as was the case with most glucose-lowering agents—SGLT2 inhibitors would have absolutely no additional benefit other than their glucose-lowering effect, and others even expressed their concerns about the potential contribution of the long-term usage of SGLT2 inhibitors to the development of renal tubular dysfunction and even acute kidney injury (AKI) [24]. However, contrary to expectations, allocation to an SGLT2 inhibitor was shown to reduce even the risk of AKI by 23% overall (RR, 0.77; 95% CI, 0.70 to 0.84), with similar reductions observed in patients with diabetes and without diabetes [19].
Although the success of SGLT2 inhibitors is wonderful and to be celebrated, it continues to flummox researchers how they can have beneficial effects on ailing kidneys beyond their glucose-lowering effects. Elucidating the exact mechanisms should improve the pathogenic understanding of kidney diseases and inform the future development of preventive tools [15]. Although several pleiotropic mechanisms have been postulated to be responsible for the renal benefits of SGLT2 inhibitors on kidney diseases, including relief of renal hypoxia; improvement of organellar dysfunction; inhibition of inflammation, oxidative stress, and apoptosis; and induction of autophagy [25], the most compelling mechanistic models involve the activation of tubuloglomerular feedback (TGF), because it is known that hemodynamic—as opposed to metabolic—factors are the dominant drivers of kidney diseases [26,27].
Go to :
EFFECTS OF SGLT2 INHIBHITOR ON TUBULAR REABSORPTION IN PROXIMAL NEPHRON
Are glomerulotubular balance and tubuloglomerular feedback altered in diabetic kidneys?
As much as 60% to 70% of total sodium (Na+) reabsorption occurs along the proximal convoluted tubule and proximal straight tubule, and 25% to 30% of the filtered Na+ is reabsorbed by the ascending limb of the loop of Henle [28]. Thus, approximately 90% of the filtered Na+ has been retrieved by the time the filtered fluid reaches the macula densa cells at the transition to the distal convoluted tubule [28], indicating that the proximal nephron including the proximal tubule and loop of Henle is responsible for most Na+ reabsorption in kidneys. In principle, both net Na+ reabsorption and fluid reabsorption by the proximal tubule increases with the amount of fluid available for reabsorption; i.e., the rate of filtration [28,29]. This phenomenon, which is known as glomerulotubular balance (GTB), occurs almost entirely in the proximal tubule and in the cortical thick ascending limb of Henle, which locally involve the highest O2 demand driven by the sodium–potassium ATPase (Na+ -K+ -ATPase), which provides Na+ potential to reabsorb solutes from the filtrate, ultimately resulting in a definition of the Na+ amount delivered to the juxtaglomerular apparatus [30]. Therefore, if glomerular filtration rate (GFR) increases and distal delivery decreases, this implies a primary increase in tubular reabsorption independent of GTB [29].
Despite GTB, the delivery of Na+ to the tubular segments beyond the proximal tubule and/or loop of Henle will increase whenever GFR increases or whenever a primary impairment of proximal Na+ reabsorption occurs due to transporter malfunction or dysregulation [28,31]. The amount of Na+ escaping the reabsorption in the proximal nephron is sensed by macula densa in the juxtaglomerular apparatus and sets the afferent arteriole tone, the mechanism of which is referred to as TGF [30]. The release of adenosine and adenosine triphosphate (ATP) that originated from the macula densa is involved in the constriction of the afferent arteriole [30]. Thus, TGF form a negative feedback system that stabilizes both single-nephron GFR and distal salt delivery [32]. By working in harmony, total responsibility for salt balance would be relegated downstream from the macula densa [30], leading to a reduction of Na+ reabsorption in distal nephron to less burdensome levels.
Are there any changes of GTB and TGF that occur with diabetes? In type 1 diabetic patients, the flow of water and flux of Na+ to the segments distal to the proximal tubule remains unchanged during variations of GFR [33], indicating that the GTB is preserved for glucose. Moreover, TGF is reset upward, especially in early diabetes, which can exacerbate hyperfiltration [31]. In contrast to type 1 diabetes, type 2 diabetes may have a slightly more complicated mechanistic basis for TGF. Hyperglycemia enhances Na+ reabsorption, thus decreasing the delivery in macula densa with suppressed TGF signal in both type 2 diabetes and type 1 diabetes [34]. Not unnaturally, salt load also affects proximal tubular reabsorption, glomerular hyperfiltration, and intrarenal angiotensin concentration [35]. In type 2 diabetes, the expressions and functions of connexins such as Cx37, Cx40, and Cx43 in juxtaglomerular apparatus are disturbed, and these connexin abnormalities appear to induce the removal of TGF signal, which dilates afferent arterioles and activates the renin-angiotensin system [34,36]. When salt intake is increased in subjects with type 1 diabetes, salt load reduces proximal tubular reabsorption, and as a result the sodium chloride (NaCl) delivery to macula densa is increased, which restores TGF signals to produce adenosine that constricts afferent arterioles and ameliorates glomerular hyperfiltration, thereby ameliorating albuminuria. This is called the ‘salt paradox’ [34], and some researchers have claimed that this anomaly could account for the positive association between salt intake and kidney survival reported in the FinnDiane study, because proximal salt reabsorption appears to be more sensitive to inhibition by dietary salt loading in the presence of diabetes than in its absence [27,37]. Despite some debates, similar findings have been obtained in patients with type 2 diabetes [38], suggesting that multiple factors—including hyperglycemia, gap junction malfunction, and the amount of salt intake—in type 2 diabetes would determine TGF response. Collectively, although TGF may operate in different ways according to the specific types of diabetes, it seems apparent that GTB and TGF affect the progression of diabetic kidney disease [39].
How do SGLT2 inhibitors affect glomerulotubular balance and tubuloglomerular feedback?
SGLT2 is expressed in the S1/S2 segment of the proximal tubule, while sodium-glucose cotransporter-1 (SGLT1) is located in the S3 segment of the proximal tubule [40]. Since Na+ -dependent cotransporters linked to glucose, phosphate, amino acids, lactate, and other molecules only make small contributions to total Na+ uptake in apical membrane, glucose-coupled Na+ absorption accounts for <10% of the total Na+ reabsorption in the proximal tubule, while SGLT2 mediates less than 5% of Na+ uptake along the proximal tubule, given a glucose reabsorption of 120 to 180 g of glucose per day as well as the stoichiometry of the SGLTs [28,40]. This indicates that SGLT2 only accounts for a small percentage of total Na+ reabsorption along the nephron, therefore indicating that SGLT2 inhibition would make no difference in GTB or TGF in normal kidneys. However, in diabetes, with exposure to high glucose, an increase in SGLT2 mRNA levels of 38% to 56% is observed in kidneys [41]. It has been reported that both the mRNA and protein expressions of SGLT2 are increased in kidneys biopsied from human subjects with diabetic nephropathy, even in advanced kidney disease [42], and SGLT1 and SGLT2 activities in diabetic condition may account for more than 14% of renal Na+ reabsorption along the whole tubule [22], which is convincing evidence for the role of SGLT2 inhibitors in the treatment of diabetic kidney disease. By contrast, treatment with SGLT2 inhibitors has been shown to fail to modify renal hemodynamic function or attenuate proteinuria in small studies and reduce the primary composite renal outcome in large trials in patients with focal segmental glomerulosclerosis [19,43,44], suggesting that the increased abundance of SGLT2 would be an essential prerequisite for the effectiveness of SGLT2 inhibitors.
The inhibition of SGLT2, which is typically upregulated in diabetic kidneys, leaves a higher-than-normal concentration of Na+ in the lumen of the proximal tubule, and after passing through the loop of Henle, the uptake of this Na+ by the macula cells exceeds the capacity of their Na+-K+-ATPase in the basolateral membrane, thus causing an increase in the intracellular Na+ concentration [45]. This in turn creates an osmotic gradient that causes the cells to swell and leak ATP across the basolateral membrane, after which ATP is converted to adenosine by an extracellular nucleotidase, and the adenosine binds to adenosine A1 receptors on vascular smooth muscle cells lining afferent glomerular arterioles [45,46]. This finally leads to SGLT2-inhibitor-mediated TGF response by causing afferent arteriolar constriction [46], which may make GTB stable. Exploratory research has demonstrated that 8 weeks of treatment with empagliflozin in patients with poorly controlled type 1 diabetes decreased renal plasma flow and attenuated hyperfiltration in association with urinary adenosine [47,48]. This is clinically presented by an acute drop or initial dip in the eGFR of 4 to 6 mL/min/1.73 m2 in the initial weeks following the initiation of SGLT2 inhibitors [49].
Is this action of SGLT2 inhibitors also observed in patients with type 2 diabetes? When dapagliflozin was administered to patients with type 2 diabetes, an acute drop in GFR caused by dapagliflozin was accompanied by a reduction in renal blood flow and renovascular resistance [50], suggesting that the acute drop in GFR may be attributed to vasodilation of the renal efferent arteriole, similar to how renin-angiotensin system blockades reduce single-nephron glomerular hypertension [49]. However, before considering all the facts, we should not jump to the conclusion that SGLT2 inhibitors reduce hyperfiltration in different ways according to the specific types of diabetes.
Do changes in renal perfusion and arteriolar resistances by SGLT2 inhibitors differ between type 1 and type 2 diabetes?
As mentioned above, some researchers have speculated that the increase in NaCl delivery following SGLT2 inhibitors leads to increased production and binding of adenosine to the adenosine A1 receptor, ultimately causing preferential vasoconstriction of the afferent arteriole in type 1 diabetes and vasodilation of the efferent arteriole in patients with type 2 diabetes [48]. This assumption is based on data from two previous studies [46,51]: one study showed that, in 27 patients with type 1 diabetes, treatment with empagliflozin for 8 weeks significantly reduced GFR by 19% and effective renal plasma flow by 31% as well as significantly increased filtration by 12% [46]. In another study examining 24 patients with type 2 diabetes, treatment with dapagliflozin for 12 weeks significantly reduced GFR by 7% but not effective renal plasma flow, leading to a 6% reduction in the filtration fraction [50,51]. Further, renal vascular resistance was increased by 37% in type 1 diabetes patients but decreased by 6% in type 2 diabetes [50]. Although the estimated intraglomerular pressure was reduced in both populations, the mechanisms by which the intraglomerular pressure decreased were different: empagliflozin increased the afferent arteriolar resistance by 66% without a significant change in the efferent arteriolar resistance in type 1 diabetes patients, while empagliflozin reduced efferent arteriolar resistance by 8% without a significant change in the afferent arteriolar resistance [46,50,51]. However, these findings might not serve as proof that the mechanism by which SGLT2 inhibition lowered GFR after treatment initiation differ according to the type of diabetes. For one, the differential effects of SGLT2 inhibitors on renal hemodynamic physiology between studies involving patients with type 1 diabetes and type 2 diabetes could be attributable to differences in patient characteristics, including differences in treatment period and baseline renal hemodynamic status, such as age, diabetes duration, comorbidities, blood pressure, glycemic control, body mass index, and concomitant medications including renin-angiotensin system blockade, statin, and insulin [49,50]. Second, it could be argued that studies using small sample sizes of less than 30 subjects are not meant to be representative of all diabetic populations. Thus, further research is needed to investigate whether the types of diabetes by themselves could be responsible for the different renal effects of SGLT2 inhibitors [50].
Given that an increase in urinary adenosine excretion was observed after SGLT2 inhibition on both type 1 and 2 diabetes patients [50], some researchers have suggested that adenosine primarily causes afferent arteriolar constriction via stimulation of the adenosine A1 receptor in type 1 diabetes, while adenosine can induce efferent arteriolar dilatation via adenosine A2 receptor in the presence of renin-angiotensin system blockade in type 2 diabetes [48,52,53]. However, another study reported that the effect of dapagliflozin in people with type 2 diabetes on renal hemodynamic physiology was similar in subjects with and without renin-angiotensin system blockade [51]. Therefore, the effect of SGLT2 inhibitors on both afferent and efferent arterioles may be more complicated than might previously have been thought. Previous experiments have demonstrated that adenosine A1 receptors appear exclusively in the glomerular entrance segment of the afferent arteriole whereas adenosine A2 receptors appear to be expressed in low density in more proximal regions in isolated perfused rabbit afferent arterioles [53,54]. Another study has indicated that adenosine A1 and A2 receptors are expressed on the afferent and efferent arterioles of juxtamedullary nephrons, such that adenosine A2 receptor-mediated vasodilation partially buffers adenosine-induced vasoconstriction in both pre- and postglomerular segments of the renal microvasculature [55,56]. This adenosine-mediated net effect on arterioles based on the type of adenosine receptor may appear to differ depending on the nephron’s location in the kidney; responding to high adenosine concentrations, in superficial nephrons, adenosine A1 receptor-mediated afferent arteriolar constriction may dominate, while in juxtamedullary or deep cortical nephrons, adenosine A2 receptor-mediated vasodilatation may primarily occur [53]. Although both adenosine A1 and A2 receptors are abundantly expressed in the kidney, the net effect of adenosine in the kidney is to constrict [57]. Since low extracellular adenosine concentrations and a secondary upregulation of adenosine A1 receptors in afferent arterioles are observed in early diabetic condition [58], adenosine A1 receptor-mediated superficial cortical vasoconstriction by SGLT2 inhibition in patients with diabetes would be expected to prevail, with rather weak juxtaglomerular cortical vasodilation via adenosine A2 receptor. No matter which arteriole is more affected by SGLT2 inhibitors, the final effect of SGLT2 inhibition in diabetic kidneys is a reduction in intraglomerular pressure (Fig. 1).
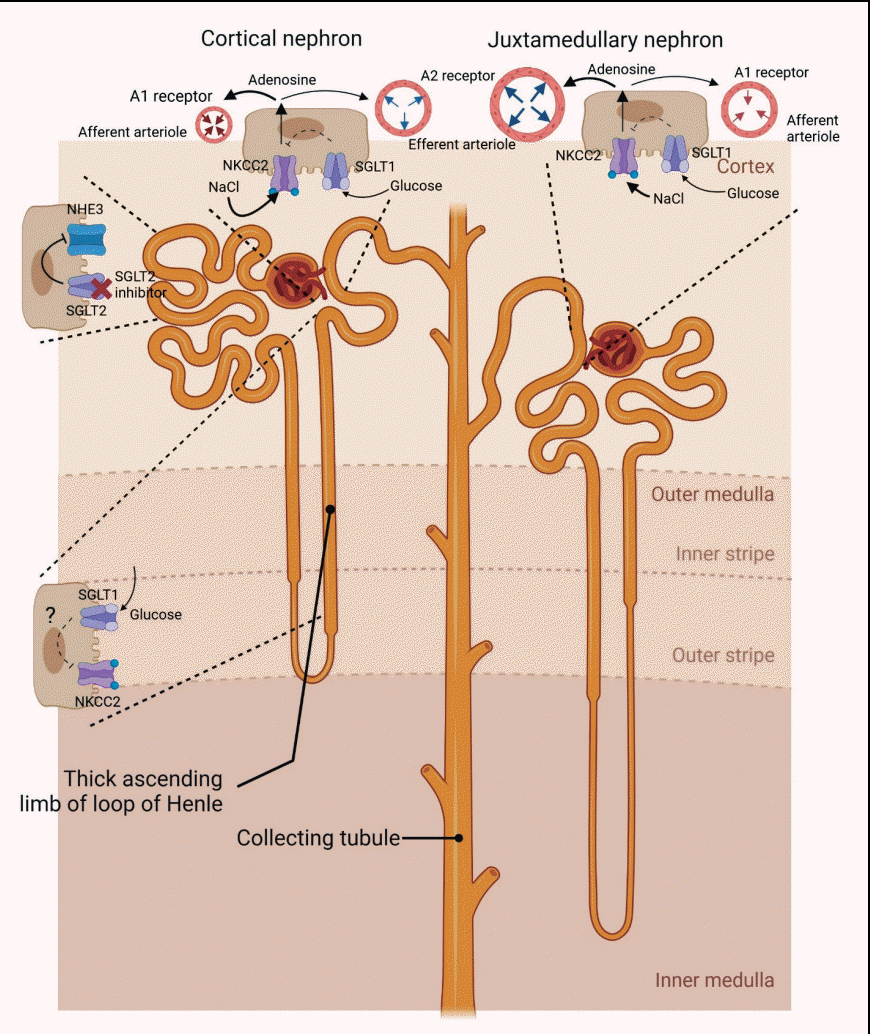 | Fig. 1.Proposed effects of sodium-glucose contrasnporter-2 (SGLT2) inhibitors on tubular transports in proximal nephron. In diabetic condition, the expression of SGLT2 is increased with exposure to the increased glycosuria, and then SGLT2 in diabetes accounts for more Na reabsorption than usual in normal status. SGLT2 inhibitors increase the distal delivery of sodium chloride (NaCl) by inhibiting SGLT2 as well as decreasing sodium–hydrogen exchanger 3 (NHE3) activity (which may not be a specific action of SGLT2 inhibitors). Sodium-glucose contrasnporter-1 (SGLT1) is expressed on the thick ascending limb of the loop of Henle, the macula densa, and the proximal tubule. The increased luminal glucose resulting from SGLT2 inhibition may be reabsorbed via SGLT1 on the thick ascending limb and the macula densa, and it may then repress upregulation of Na+-K+-2Cl− cotransporter (NKCC2). In macula densa, the increase in luminal NaCl concentration is sensed by NKCC2, causing the release of adenosine from macula densa, and then affecting arterioles. This process may be subject to check by SGLT1 activity stimulated by the increased luminal glucose concentration. The adenosine released from macula densa can act as a vasoconstrictor in afferent arteriole and vasodilator in efferent arteriole by binding A1 receptor and A2 receptor respectively. Responding to high adenosine concentrations, adenosine A1 receptor-mediated afferent arteriolar constriction may dominate in cortical nephrons, while adenosine A2 receptor-mediated vasodilatation may be more dominant in juxtamedullary or deep cortical nephrons. Under any location of the nephron, the final outcome of SGLT2 inhibitors is a reduction of intraglomerular pressure.
|
Could SGLT2 inhibitor sustainably activate tubuloglomerular feedback system?
If SGLT2 inhibitors continuously decrease solute reabsorption in the proximal tubule, it would be possible to sustainably increase Na+ delivery to macula densa. However, after a given period of time, partial or even complete compensation in reabsorption in segments proximal to the macula densa or resetting or saturation of the TGF system could offset the initial response of SGLT2 inhibitors [31]. From the viewpoint of diuresis and natriuresis, an eight-week treatment with empagliflozin did not significantly increase 24-hour urine volume in subject with renal normofiltration but caused a significant rise in 24-hour urine volume in subjects with renal hyperfiltration [46]; meanwhile, in patients with type 2 diabetes, 28 days of empagliflozin therapy did not cause clinically relevant and significant changes in urine volume despite a trend of an increase in 24-hour urine volume even after day 1 of administration [59]. It was also reported that the administration of canagliflozin led to an increase in urinary Na+ excretion on day 1 with a return to baseline afterward [60]. Based on these findings, we might guess that the increased NaCl delivery to macula densa by SGLT2 inhibition could be weakened in time by an increase in the function of SGLT1 in the proximal tubule as well as an increase in reabsorption by Na+-K+-2Cl− cotransporter (NKCC2) in loop of Henle (Fig. 1) [60].
Similar examples include acetazolamide, which is a carbonic anhydrase inhibitor, and furosemide, which is a loop diuretic, both of which are diuretics that work in the proximal nephron. Acetazolamide reduces proximal tubular reabsorption inhibiting bicarbonate reabsorption via decreasing carbonic anhydrase activity, ultimately resulting in an initial increase in macula densa solute delivery and subsequent activation of TGF [31]. However, TGF response by acetazolamide does not remain activated through resetting of the turning point of the TGF curve, possibly due to increased nitric oxide (NO) levels [31,61]. Furosemide reduces Na+ reabsorption in the thick ascending limb of the loop of Henle by inhibiting NKCC2, which initially increases the solute concentration of fluid reaching the macula densa and activates TGF response [31]. Conversely, furosemide can also block the TGF system by inhibiting the sensing of the NaCl concentration in the macula densa [62]. Although it remains unclear whether tubular concentrations achieved by the pharmacological use of furosemide tip the balance between these opposing forces toward an attenuated or activated TGF response [62], there is little evidence to date regarding loop diuretics to support their anti-proteinuric or renoprotective effects [63], so furosemide might not have a sustained effect on TGF activation.
Contrary to the existing diuretics, SGLT2 inhibitors appear to sustainably activate the TGF system. Despite the limited data on the long-term effect of SGLT2 inhibitors on the TGF system, the sustained TGF response has been speculated to be approximately 50% of the acute response [29,31,64]. There are several potential mechanisms that could be proposed to explain the sustained TGF activation of SGLT2 inhibitors. First, the inhibition of SGLT2 in diabetic rats does not appear to upregulate NKCC2, as it instead causes a trend toward a decrease in NKCC2 expression [65,66], which means that—against our expectations— there is no compensatory increase in solute reabsorption in the loop of Henle. Interestingly, it is known that SGLT1 is also expressed on the apical membrane of the thick ascending limb and the macula densa, besides the proximal tubule [67]. In macula densa, the glucose load transported from tubular fluid via SGLT1 seems to activate the cyclic adenosine monophosphate/protein kinase A and phosphoinositide 3-kinase/protein kinase B (Akt) signaling pathways, which would upregulate the expression and phosphorylation levels of NO synthase and then increase NO [67]. Although the functional significance of SGLT1 located in the apical membrane of the ascending limb of the loop of Henle downstream of macula densa still remains unclear [68], similar to macula densa, the activated SGLT1 in the thick ascending limb under the condition of an increase in luminal glucose with SGLT2 inhibitors may suppress NKCC2, resulting in sustained NaCl delivery to macula densa. However, there is another point to consider in terms of this hypothesis: although the decreased expression of NKCC2 in the thick ascending limb of loop of Henle—which is inhibited by SGLT1 activated with diabetic condition and SGLT2 inhibition—could lead to a sustained NaCl delivery to macula densa, SGLT1 in macula densa, which may also be activated in the same way, would hinder the increased luminal NaCl concentration from being sensed by NKCC2 on the apical membrane of macula densa (Fig. 1). Altogether, glucose concentration sensed by SGLT1 located on the luminal side of macula densa cells upregulates neuronal NO synthase, which may inhibit the TGF response [48]. Second, SGLT2 inhibitors induce the suppression of sodium–hydrogen exchanger 3 (NHE3) in the proximal tubule, which may explain the continuous NaCl delivery to macula densa [69,70]. SGLT2 and NHE3 are interlinked by membrane associate protein 17 (MAP17) that interacts via postsynaptic density protein 95/tight junction protein 1 (PDZK1), indicating that SGLT2 increases the activity of NHE3 [70]. Since NHE3 expression is enhanced in diabetes, SGLT2 inhibitors may maintain distal NaCl delivery reaching the macula densa by indirectly inhibiting NHE3 (Fig. 1). However, this is an issue that requires additional consideration. Beyond SGLT2 inhibitors, lixisenatide, which is a glucagon-like peptide receptor 1 agonist, inhibited upregulation of NHE expression, and voglibose, an α-glucosidase inhibitor, even decreased NHE3 expression in diabetic rat kidneys [66]. Therefore, the effect on NHE3 expression might not be limited to SGLT2 inhibitors. Meanwhile, the fact that SGLT2 inhibitors inhibit NHE3 could be related to the possibility that bicarbonate (HCO3-) reabsorption is decreased in the proximal tubule and then causes metabolic acidosis [48,70]. Fortunately, the finding that SGLT2 inhibitors increase ammonia excretion may neutralize fears of the development of clinically significant acidosis after the initiation of SGLT2 inhibitors [70]. Only dedicated studies specifically investigating the impact of SGLT2 inhibitors on the proximal nephron reabsorption and TGF system will provide us with satisfying answers.
Go to :
EFFECTS OF SGLT2 INHIBHITOR ON TUBULAR REABSORPTION IN DISTAL NEPHRON
Does diabetes affect the distal tubular reabsorption of salt?
The distal nephron of the kidney, including the distal convoluted tubule and the collecting duct, plays an important role in the fine tuning of Na+ reabsorption [71]. Some studies using rodent models have reported increased expression of sodium chloride cotransporter (NCC) located in the distal convoluted tubule under diabetic conditions [72,73], while others have shown that the abundance of NCC was not increased in Otsuka Long-Evans Tokushima Fatty (OLETF) rats [71]. Meanwhile, in another study using OLETF rats, immunostaining demonstrated that NCC was significantly increased in diabetic kidneys, but that immunoblotting for NCC failed to show the same results as immunostaining [66].
It should not be necessary to lean to one side. First, it is known that insulin or hyperinsulinemic states are associated with increased activity and phosphorylation of NCC [74,75]. Another interesting observation is that dysfunction in NCC could be one of the main causes through which insulin resistance in patients with type 2 diabetes leads to chronic hyperglycemia state and subsequently to diabetic kidney disease risk [76,77]. This may support the possible link between diabetes and Gitelman syndrome caused by the mutation of solute carrier family 12 member 3 (SLC12A3) gene encoding the thiazide-sensitive NCC [77]. These conflicting results suggest that there are factors other than diabetic condition in which NCC activity is involved.
Similar to NCC, the epithelial sodium channel (ENaC), which is primarily expressed in the apical membrane of the epithelial cells lining the distal segment of nephrons, is activated in diabetic patients with nephropathy [78]. It has also been found that high glucose stimulates ENaC expression in human cortical collecting duct cells [79]. A previous study has found no significant difference in the expressions of ENaC α- and γ-subunits between Long-Evans Tokushima Otsuka (LETO) and OLETF rats [66], while another study has suggested that increased ENaC activity stimulated by insulin from glucose-induced hyperinsulinemia following a meal would be a more important mechanism to preserve Na+ [80]. Typically, Na+ transport in the connecting tubule, cortical collecting duct, and inner medullary collecting duct are known to be stimulated modestly by aldosterone [28]. Because angiotensinogen and ACEs are present in the distal nephron lumen, luminal renin, which is synthesized and secreted into the tubule lumen by collecting duct cells, may also stimulate luminal angiotensin II formation, thus leading to enhanced ENaC activity [81]. This renin production in the collecting duct is markedly increased in the context of diabetes as well as by circulating angiotensin II [81].
Could SGLT2 inhibition lead to altered Na+ reabsorption in distal nephron segments?
Treatment with empagliflozin for 12 weeks in OLETF rats did not significantly change NCC expression compared to untreated diabetic rats [66]. Meanwhile, another study showed that administration of ipragliflozin—another SGLT2 inhibitor—for one week reduced NCC levels in the kidney of db/db mice [82]. It has been reported that hyperglycemia and activation of the glycolytic pathway result in the accumulation of intermediate metabolites that stimulate de novo diacylglycerol synthesis and PKC activation [82,83]. This PKC activation then seems to phosphorylate and inactivate the ubiquitin ligase component Kelch-like 3 (KLH3) in the distal convoluted tubule, ultimately resulting in increased NCC activity and subsequent NaCl retention [82]. Interestingly, PKC activity, the phosphorylation of serine 433 (S433) in KLHL3 (KLHL3S433-P), and NCC levels in the diabetic mouse kidney could all be lowered by ipragliflozin but not by pioglitazone, although both agents effectively reduced blood glucose [82]. The precise mechanism by which SGLT2 inhibition causes a decrease in NCC has yet to be identified, but these observations may be attributable to the mechanical difference between SGLT2 inhibitors and insulin sensitizers in reducing blood glucose. Since altered functions of the NCC have been shown to have profound effects on blood pressure regulation, as represented by the excessive activation and inactivation of the NCC, such as Gordon’s and Gitelman syndromes, respectively, a close association between Na+ handling by NCC and blood pressure appears to be important [84]. Therefore, SGLT2 inhibitors may have a role in blood pressure regulation through possible modulation of NCC, although there is a need for further research to explore the possible direct or indirect effect of SGLT2 inhibitors on NCC (Fig. 2).
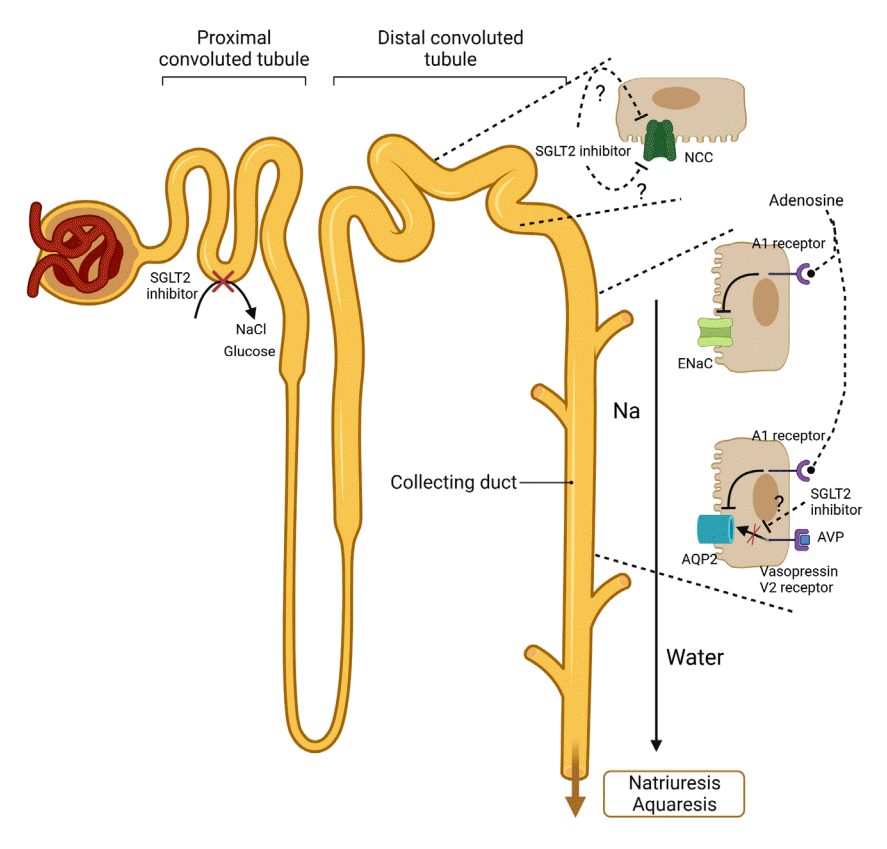 | Fig. 2.Proposed effects of sodium-glucose contrasnporter-2 (SGLT2) inhibitors on tubular transports in distal nephron. The treatment with SGLT2 inhibitors cause a decrease in sodium chloride cotransporter (NCC) levels by not-yet-identified mechanisms. Despite some conflicting results, the expression epithelial sodium channel (ENaC) γ-subunit is decreased by SGLT2 inhibitors. The interstitial adenosine, released from macula densa, could inhibit ENaC activity by activating adenosine A1 receptors in the collecting duct. The increased interstitial adenosine could also suppress aquaporin-2 (AQP2) in collecting duct by binding the adenosine A1 receptor and disrupting signaling pathways between vasopressin V2 receptor and AQP2 synthesis. Collectively, disturbed Na reabsorption in various tubules and decreased water reabsorption in distal nephron by SGLT2 inhibitors could lead to natriuresis and aquaresis. AVP, arginine vasopressin.
|
In high-fat diet diabetic mice, dapagliflozin administration for 18 days has been shown to increase the expression of ENaC α-subunit by 29% while decreasing the expressions of ENaC β- and γ-subunit by 13% and 26%, respectively, compared with to-control mice [85]. On the other hand, diabetic rats treated with empagliflozin for 12 weeks displayed decreased renal protein expressions of ENaC α- and γ-subunits compared to untreated OLETF rats [66]. Given that gain-of-function mutations in the ENaC α-, β-, or γ-subunit have been associated with Liddle syndrome, as characterized by hypertension, suppressed aldosterone, and hypokalemia, ENaC activity may play a role in salt-sensitive hypertension [86,87]. Among ENaC subunits, there has been a series of results suggesting that ENaC γ-subunit plays a more potent role than ENaC β-subunit in assembling and trafficking the complex to the surface, although both ENaC β- and γ-subunits are essential in the translocation of the channel complex to the plasma membrane [88]. As a result, the SGLT2 inhibitors-induced decrease in the expression of ENaC γ-subunit, the pivotal subunit of ENaC, could contribute to natriuresis.
The other scenario wherein SGLT2 inhibitors could inhibit salt reabsorption in distal nephron is that renal interstitial adenosine—which are, as mentioned above, increased by SGLT2 inhibition—could activate adenosine A1 receptors in the collecting duct and then inhibit ENaC activity (Fig. 2). Adenosine action on its receptor is known to inhibit ENaC [81]. There is a need for further experiments to investigate the detailed mechanisms mediating adenosine inhibition of ENaC by SGLT2 inhibitors.
How could SGLT2 inhibitors increase excretion of free water?
Recently, two randomized, double-blind, placebo-controlled trials provided evidence that empagliflozin is an effective treatment in both hospitalized patients with syndrome of inappropriate antidiuresis (SIAD) and outpatients with chronic SIAD [89,90]. Both euvolemic and hypervolemic hyponatremia primarily result from arginine vasopressin (AVP)–mediated free water retention, which is represented by decreased free water clearance [91,92]. Fluid restriction or vasopressin V2 receptor antagonists are typically applied to treat hyponatremia [91]. However, fluid restriction has limited efficacy, and vasopressin V2 receptor antagonists, called vaptans, may overcorrect serum Na+ [91,92]. SGLT2 inhibitors increase glucosuria, which leads to osmotic diuresis, and this may result in increased excretion of free water [92]. This action by SGLT2 inhibitors could help treat SIAD slowly without major adverse events. The question here is, can the increased free water clearance be explained by glucosuria and subsequent osmotic diuresis alone? In type 2 diabetic Goto-Kakizaki (GK) rats, treatment with ipragliflozin for 8 weeks has been shown to increase osmolar clearance, solute‐free water reabsorption, and electrolyte‐free water clearance [93]. That study showed that ipragliflozin increased AVP levels, the renal expression of the vasopressin V2 receptor, and the phosphorylation of aquaporin-2 (AQP2) at Ser269 [93]. By contrast, more long-term use of empagliflozin in OLETF rats led to higher electrolyte-free water clearance, a decrease in total AQP2 expression, and an increase in the phosphorylation of AQP2 at S261 through the activation of p38-mitogen-activated protein kinase (MAPK), protein phosphatase 2B (PP2B) and glycogen synthase kinase 3α (GSK3α), and cyclin-dependent kinases 1 and 5 (cdk1 and cdk5), regardless of the upregulation of vasopressin V2 receptor [66]. Although previous studies have shown conflicting results, the SGLT2 inhibitor-induced inappropriate AQP2 downregulation resulting from a disruption of signaling between vasopressin V2 receptor and AQP2 synthesis may be fairer. As with ENaC, adenosine can also inhibit AQP2 in the collecting duct via the adenosine A1 receptor [81]. Finally, SGLT2 inhibition could cause aquaresis by inhibiting AQP2 (Fig. 2).
Go to :
CONCLUSIONS
In summary, the SGLT2 inhibition leads to an increase in NaCl delivery to the macula densa, resulting in enhanced production and binding of adenosine to adenosine receptors. Consequently, adenosine A1 receptor-mediated constriction of the afferent arterioles may predominate in cortical nephrons, while adenosine A2 receptor-mediated vasodilation may be more pronounced in juxtamedullary or deep cortical nephrons. Several potential mechanisms may explain the sustained activation of the TGF by SGLT2 inhibitors, including the absence of changes in NKCC2 and the suppression of NHE3. In the distal nephron, SGLT2 inhibitors seems to inhibit NCC, ENaC and AQP2 through mechanisms that have not yet been fully identified but may involve adenosine-adenosine receptor interactions. These interactions may contribute to continuous natriuresis and aquaresis.
With the aging of the population and the still-increasing prevalence of causative diseases, kidney disease has persisted as an unseen threat continually coming from many directions. Over the last 5 years, numerous advancements emerged in the management of CKD with diabetes [94]. Notably, SGLT2 inhibitors have become indispensable for the treatment of CKD with or even without diabetes [95]. In ‘the new era of SGLT2 inhibitors,’ it is likely that almost all CKD patients should be put on SGLT2 inhibitors. This ‘change agent’ is expected to continue to make a difference in the lives of numerous patients with CKD.
However, it is important that we do not allow ourselves to be carried away by the success of SGLT2 inhibitors. There are still so many questions to resolve, including whether the mechanisms underlying the renal benefit of SGLT2 inhibitors in both diabetic and non-diabetic kidney diseases are identical, how other concomitant conditions or treatments could affect the hemodynamic effects of SGLT2 inhibitors, how the actions of SGLT2 inhibitors might change in advanced CKD, and whether dual SGLT1 and SGLT2 inhibition can provide more renal advantages over SGLT2 inhibition alone [30,96]. Continuous and comprehensive research into the renoprotective mechanisms of the SGLT2 inhibitors, which could allow us to keep looking for new adventures, new directions to investigate, and new lessons to learn, will eventually lead to more success stories.
Go to :
 XML Download
XML Download