

 PDF
PDF Citation
Citation Print
Print



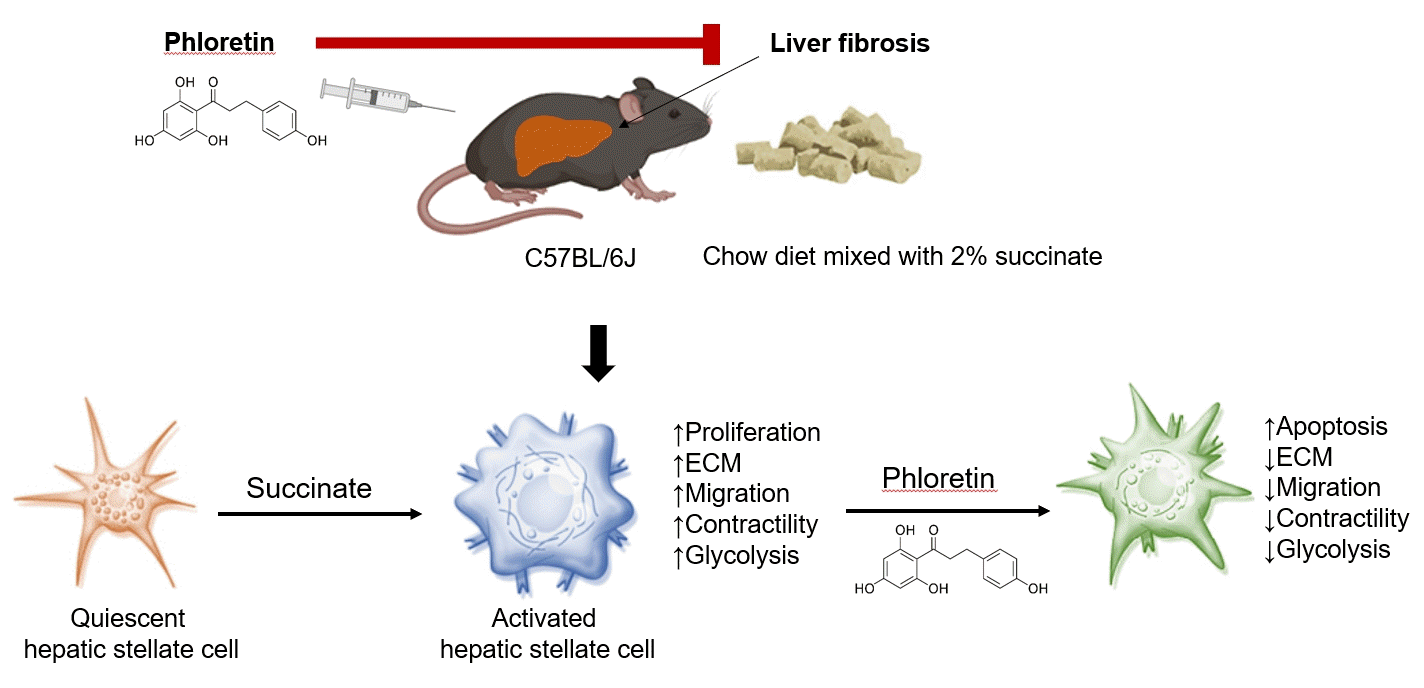
INTRODUCTION
Liver fibrosis which accumulates a tough tissue around hepatic cells is the wound-healing process to response to chronic liver injury that progress could lead to cirrhosis and liver failure [1]. Hepatic stellate cells (HSCs) play an essential role in the progression of liver fibrosis, because they are the major source of generated extracellular matrix (ECM) proteins. Following liver damage, HSCs undergo a morphological shift to myofibroblast-like cells, including increased collagen production and cell contractility [2]. At the sites of liver injury, these activated HSCs migrate and proliferate, perpetuate hepatic inflammation and play a critical role in the formation of fibrosis. Therefore, activated HSCs are potential therapeutic targets for the treatment of chronic liver diseases [3].
Succinate, known as an intermediate in the tricarboxylic acid (TCA) cycle, plays an important role in the generation of energy molecular adenosine triphosphate (ATP) in the mitochondria. In conditions of oxygen deprivation, succinate may accumulate and be secreted in the extracellular area. This time, succinate appears to be a ligand by binding to its receptor in the cellular plasma membrane and transmits signal into the target cells. Succinate and its receptor have been linked to several pathological processes such as hypertension [4,5], diabetes and obesity [6-9]. Our team demonstrated that succinate binds to its receptor in HSCs and induces their activation by promoting α-smooth muscle actin (α-SMA) and ECM protein production, increasing proliferation, and migration, and inhibiting apoptosis of cells in in vitro studies [10,11]. In vivo, mice consumed succinate every day for 16 weeks could induce liver fibrosis and increase triglyceride accumulation [12].
It also has been well established that aerobic glycolysis, known as the Warburg effect, is a hallmark of cancer cells metabolism [13]. Glycolysis is a faster and shorter pathway to generate ATPs in cancer cells, and is commonly employed by some type of cells to meet the high metabolic demands during rapid proliferation [14]. Recent evidence suggests that aerobic glycolysis is a characteristic metabolic phenotype of activated HSCs during hepatic fibrosis, and activated HSCs behave similarly to proliferative cancer cells in terms of bioenergetic and biosynthetic requirements [15], suggesting the importance of aerobic glycolysis in regulating the activation, proliferation, and contraction of HSCs.
Phloretin, a phenol-based antioxidant expressed in high levels in pears, apple tree leaves, and strawberries, can combat obesity caused by poor diets and certain metabolic disorders [16]. Previous studies showed that oral administration of phloretin improves dyslipidemia in diabetic rats [17]. Another study [18] have shown that the expression of certain genes associated with lipogenesis and triglyceride storage, such as acyl-coenzyme A (CoA) synthetase long-chain family member 1 (ACSL1), glucose transporter 4 (Glut-4), and phosphoenolpyruvate carboxyl kinase 1 (PEPCK1), are also regulated by phloretin. Shu and colleagues reported that phloretin increases concentrations of non-esterified fatty acids, enhances glucose utilization, and decreases lactate production in porcine primary adipocytes [19]. In addition, phloretin has been found to inhibit lipopolysaccharide-induced activation of macrophages [20], and to prevent high-fat diet-induced obesity and carbon tetrachloride-induced liver injury in mice [21,22]. In this study, we evaluated whether phloretin had therapeutic potential in treating hepatic fibrosis in a mouse model of sodium succinate diet-induced model of liver fibrosis. Furthermore, we also aimed to elucidate the role of aerobic glycolysis in the activation of HSCs and to discover the molecular mechanisms through which phloretin can reduce succinate-induced HSCs activation.
Go to : 
METHODS
Materials
Succinic acid from Sigma (St. Louis, MO, USA); phloretin was supplied by Tokyo Chemical Industry Co. Ltd. (Tokyo, Japan).
Cell cultures
LX-2 cells are known as human immortalized HSCs. LX-2 cells were maintained in a humidified 37°C incubator with 5% carbon dioxide (CO2) and were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with fetal bovine serum (FBS; 10%) and penicillin/streptomycin (1%).
Animal experiments
The animal research protocol was approved by the Institutional Animal Care and Use Committee of Kangwon University (KW-160601-1). Male C57BJ6 mice weighing 22 to 25 g (6 to 8 weeks old) were provided by Doo Yeol Biotech (Seoul, Korea). All mice were housed at a temperature of 22°C±1°C, with free access to water and food, on a cycle of 12/12-hour light/dark. The mice were randomly divided into three groups and treated for 16 weeks; these groups included a control group (chow diet-fed mice+intraperitoneal injection with vehicle dimethyl sulfoxide [DMSO], n=7), a sodium succinate group (2% sodium succinate mixed with chow diet+intraperitoneal injection with vehicle DMSO, n=7), and a sodium succinate+phloretin group (2% sodium succinate mixed with chow diet+intraperitoneal injection of 10 mg/kg phloretin every other day, n=5). The dose of phloretin was used in this research based on previous mice model of obesity, liver cancer, lung fibrosis, asthma [21,23-25]. After 16 weeks, mice were euthanized with CO2 and tissue was collected and lysed for analysis.
Real-time polymerase chain reaction
RNeasy Plus Micro Kit (REF 74234, Qiagen, Hilden, Germany) was used to isolate total ribonucleic acid (RNA) in LX-2 cells, which was then transcribed to coding deoxyribonucleic acid (cDNA) using oligo dT (REF C110A, Promega, Madison, WI, USA) and hypescript RT master mix 601-710 (contain hyperscript reverse transcriptase, mixture deoxynucleotide triphosphates (dNTPs), reaction buffer, stabilizer, RNase inhibitor provided by GeneAll, Singapore). Subsequently, the mRNA copy number of several markers was determined using a real-time polymerase chain reaction (PCR) Quant Studio 6 Flex (Life Technologies, Carlsbad, CA, USA) system with SYBR Green. After PCR, a melting curve analysis was performed to assess the specificity of the PCR product as a single peak. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used for the normalization as a reference gene. The difference in the quantification cycle values for GAPDH and the respective gene was used to calculate the level of α-SMA, collagen 1, transforming growth factor β1 (TGFβ1), glucose transporter 1 (GLUT-1), and lactate dehydrogenase A (LDHA). Primers had the following 5’-3’ sequences: human α-SMA (forward: TTATGTAGCTCTG GACTT, reverse: CAACTCGTAACTCTTCTC), human collagen 1 (forward: AGAAGAACTGGTACATCA, reverse: CAT ACTCGAACTGGAATC), human TGFβ1 (forward: GCACTGTTGAAGTGCCTTAC, reverse: AGGAACTCCCCTTAACCT), human GLUT-1 (forward: TATGTGGAGCAACTGTGT, reverse: TGAAGTAGGTGAAGATGAAGA), human LDHA (forward: GATGATGTCTTCCTTAGTGTT, reverse: AGTCAGAGTCACCTTCAC).
Western blot analysis
Cells and liver tissues were lysed by WSE-7420 EzRIPA lysis kit (ATTA, Tokyo, Japan) containing radioimmunoprecipitation assay (RIPA) buffer, protease inhibitor and phosphatase inhibitor. Samples were run on sodium dodecyl sulfate-polyacrylamide gel electrophoresis and then use polyvinylidene difluoride membranes for electro-transferred. Samples were blocked for 1 hour in nonfat dry milk (5%) (BD, Sparks Glencoe, MD, USA) at room temperature and were subsequently immunoblotted with respective antibodies over night at 4℃. The membranes were incubated with secondary antibodies conjugated to horseradish peroxidase for 1 hour at room temperature, then use Westsave Star Detection Reagent System (Abfrontier, Seoul, Korea) to detect protein bands. The image of protein bands was taken by chemidoc imaging system (Bio Rad, Hercules, CA, USA). GAPDH was used as control for densitometric analysis of the respective bands, which was performed using image J software (National Institutes of Health, Bethesda, MD, USA). Primary antibodies included those specific for GAPDH (GTX627408, GeneTex, Irvine, CA, USA; dilution factor 1:5,000), α-SMA (ab5694, Abcam, Cambridge, UK; dilution factor 1:500), collagen type 1 (SAB 1402151, Sigma Aldrich, St. Louis, MO, USA; dilution factor 1:500), tissue inhibitor of metalloproteases 1 (TIMP-1; sc-5538, Santa Cruz Biotechnology, Santa Cruz, CA, USA; dilution factor 1:250), GLUT-1 (ab15309, Abcam; dilution factor 1:500), phosphorylated adenosine monophosphate protein kinase α (p-AMPKα; Thr172 #2535, dilution factor 1:1,000), AMPK (#2532, dilution factor 1:1,000), LDHA (C4B5 #3582, dilution factor 1:2,000), hexokinase II (HK II; C64G5 #2867, dilution factor 1:1,000), phosphorylated myosin light chain 2 (p-MLC2 T18/S19 #3674, dilution factor 1:1,000), MLC2 (D18E2 #8505, dilution factor 1:1,000), cleaved poly-adenosine diphosphate-ribose polymerase (PARP #9541, dilution factor 1:1,000), and PARP (#9542, dilution factor 1:1,000) were all obtained from Cell Signaling Technology, Danvers, MA, USA.
Histological analysis
Liver samples of mouse were fixed in phosphate buffered formalin (10% wt/vol) for 18 to 20 hours. The tissues were embedded in paraffin wax and serial frontal sections were cut on microtome (5 μm of thickness). Paraffin sections were deparaffinized twice for 30 minutes each by xylene, hydrated by alcohol (twice for 5 minutes each with 100% ethanol and 5 minutes each with 95%, 85%, and 75% ethanol) and rinsed for 1 minute in distilled water and then stained with Masson’s trichrome or hematoxylin and eosin (H&E), then checked at 20× magnification by Olympus microscopy (Model TH4-200, Olympus, Tokyo, Japan).
Cell proliferation assay
Cell viability was assessed using 3-(4,5-dimethylthiazol-2-yl) -2,5-diphenyltetrazolium bromide (MTT). LX-2 cells were plated in 24-well plates (25,000 cells/well) and treated with succinate (1,600 μM) with or without phloretin (100 μM) for 24 hours. MTT (1 mg/mL) in DMEM without phenol red (Welgene, Gyeongsan, Korea) was added to each well. The plates were incubated for 4 hours at 37℃ to form formazan, which was dissolve in isopropanol before measure absorbance at 570 nm using a spectrophotometer (Spectra Max 190, Molecular Devices, San Jose, CA, USA). The number of calculated cells before treatment are used as value 100% of cell proliferation.
Wound healing assay
LX-2 cells were seeded in six-well plates at a density of 5×105 cells/well in DMEM for the wound migration assay. LX-2 cells were incubated in mitomycin C (1 mg/L) for 1 hour to allow cells to reach 90% confluence. A pipette tip (200 μL, Axygen, Union City, CA, USA) was then used to scratch the injury line and the monolayers of the LX-2 cells were rinsed twice with phosphate buffered saline (1X, pH 7.4±0.1) for 10 seconds, then treated with succinate (1,600 μM) for 16 hours in the presence or absence of phloretin (100 μM) in DMEM containing 100 mL/L FBS. The image of injury lines in six-well plates was taken by Olympus microscopy (Model TH4-200, Olympus) at 0 hour, 16 hours after treatment. The widths of injury line were measure by software (cell sense standard, Olympus) and plotted against distance moved to infer cell migration.
Collagen gel contraction assay
The assay kit of collagen gel contraction was provided by Cell Biolabs Inc. (San Diego, CA, USA). The collagen gel was prepared in 24-well plates. The mixed solution of LX-2 cells (density of 1×105 cells/well) and collagen working solution (500 µL) were added to respective plate, then plates were incubated at 37℃ for 1 hour to allow gelatinization. A 1,000 µL of medium was seeded on the gel and incubated overnight at 4℃. Cells were treated with succinate (1,600 μM), with or without phloretin (100 μM) for 24 hours at 37℃. A spatula was used to gently detach the gel from the plates, then the areas of the gels were measured by software (cell sense standard, Olympus).
L-lactate colorimetric assay kit
L-lactate concentrations were quantified using a commercially available assay kit (colorimetric, ab65331, Abcam), which uses an assay protocol where lactate is oxidized by lactate dehydrogenase to form a product that produces a colored product when it interacts with a probe molecule. In particular, samples were incubated with reaction mix (lactate assay buffer, lactate enzyme mix, lactate substrate mix) for 30minutes at room temperature and then read absorbance at the wavelength 450nm by microplate reader (Spectra Max 190).
Triglyceride assays
A commercially available assay kit was used to quantify triglyceride levels (AM 157S-K, Asan Pharmaceutical, Seoul, Korea). In this assay, triglycerides are converted to free fatty acids and glycerol, which is then oxidized to form a product that reacts with a probe to produce a colored product whose concentration can be determined by spectrophotometry. In particular, samples were incubated with enzyme mix at 37℃ for 10 minutes before measured absorbance at a wavelength of 550 nm using microplate reader (Spectra Max 190).
Alanine transaminase activity assays
Alanine transaminase (ALT) activity was assayed using a commercially available kit (AM 102, Asan Pharmaceutical). ALT catalyzes the reaction between alanine and α-ketoglutaric acid to produce pyruvic acid and glutamate. Pyruvic acid levels were detected by a reaction that converts a nearly colorless probe to a colored product, and ALT activity was inferred by measuring absorbances at 505 nm using a spectrophotometer (Spectra Max 190).
Glycolytic extracellular acidification rate analysis
LX-2 cells were seeded into XF24 cell culture microplates (Seahorse Bioscience, North Billerica, MA, USA) and treated with succinate 1,600 or 400 μM with or without phloretin (50 and 100 μM), and 24 hours later, extracellular acidification rates (ECARs) were quantified using Agilent Seahorse XF analyzer according to the manufacturer’s protocol. For glycolysis assessments, the XF Glycolysis Stress Test Kit was used according to the manufacturer’s protocol.
Statistical analyses
All data were collected from at least three independent experiments. Results are expressed as means±standard error of the mean. Statistical significance in between the experiments was determined by comparing two groups using two-tailed Student’s t test. Statistical analyses were performed using Microsoft Excel (Microsoft, Redmond, WA, USA) and P values <0.05 were deemed statistically significant.
Go to :
RESULTS
Phloretin reduced the upregulation in fibrogenic markers expression, inhibited cell proliferation and increased apoptosis expression induced by succinate in HSCs
The mRNA data showed that phloretin 100 μM ameliorated significantly succinate-induced increase of fibrogenic markers compared to control (Fig. 1A). However, phloretin 50 μM didn’t show significant decrease of mRNA expression of α-SMA, collagen type I, and TGFβ1 compared to control. Succinate stimulation led to a significant increase in LX-2 cell activation, as indicated by higher levels of TGFβ1, α-SMA, and collagen type I proteins (Fig. 1B). However, when phloretin 100 μM was co-treated with succinate, it resulted in the inhibition of fibrotic protein expression more significantly compared to control (Fig. 1B).
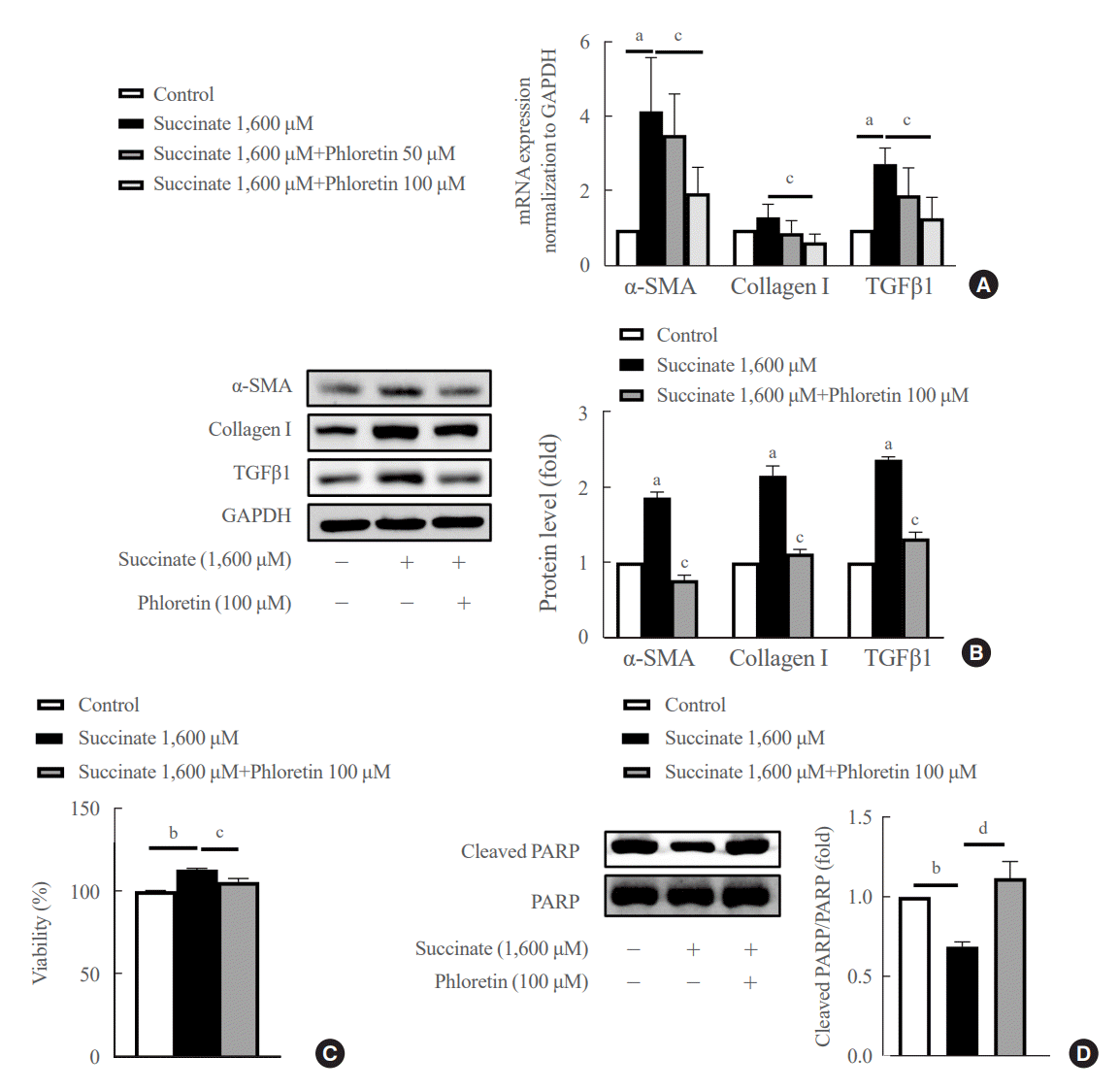 | Fig. 1.Phloretin reduced the upregulation in fibrogenic markers expression, inhibited cell proliferation and increased apoptosis of hepatic stellate cells (HSCs) induced by succinate in HSCs. LX-2 cells were treated with succinate (1,600 μM) and phloretin (50 or 100 μM) for 24 hours. (A) Real-time polymerase chain reaction to check mRNA expression of α-smooth muscle actin (α-SMA), collagen type I, transforming growth factor β1 (TGFβ1). (B) Western blot analysis of α-SMA, collagen type I, TGFβ1 were detected using specific antibodies. (C) Measure cell viability by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assessment and (D) Western blot analysis of cleaved poly-adenosine diphosphate-ribose polymerase (PARP), and PARP. Analysis of densitometry was performed and present data as the mean±standard error values of three independent experiments. GAPDH, glyceraldehyde-3-phosphate dehydrogenase. Statistical significance: aP<0.05, bP<0.001 vs. the control group; cP<0.05, dP<0.01 vs. succinate.
|
Furthermore, succinate treatment markedly increased the viability of HSCs after 24 hours, while reducing the levels of cleaved PARP, an apoptotic marker, in treated groups (Fig. 1C, D). On the other hand, phloretin was found to decrease the number of activated HSCs by increasing apoptosis of HSCs (Fig. 1D). These findings suggest that phloretin may have the potential to block HSC activation.
Phloretin inhibited succinate-induced migration and contraction of HSCs
Co-treatment with phloretin inhibited cell migration caused by succinate in LX-2 cells for 16 hours (Fig. 2A), suggesting that phloretin can inhibit cell migration processes in activated HSCs. After 24 hours of incubation, treatment of succinate reduced the collagen gel area compared to untreated LX-2 cells, whereas co-treatment with phloretin attenuated this reduction in collagen gel area (Fig. 2B). Moreover, Western blot results showed that TIMP-1 and p-MLC2 expression increased following succinate (Fig. 2C), which was attenuated by co-treatment with phloretin. Altogether, these results suggest that phloretin might inhibit the migration and contractility of activated HSCs.
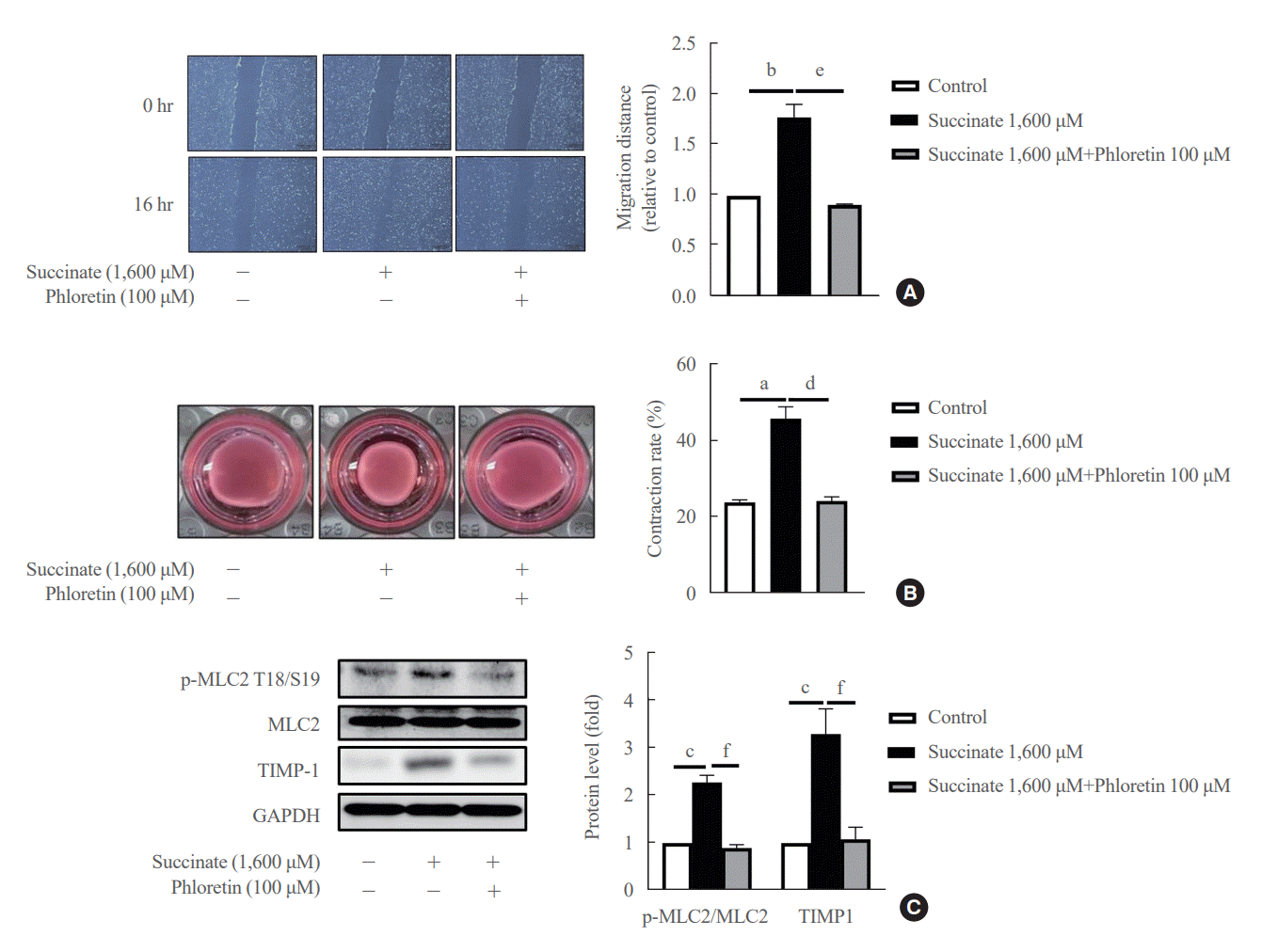 | Fig. 2.Phloretin inhibited succinate-induced migration of hepatic stellate cells (HSCs). LX-2 cells were exposure with succinate (1,600 μM) for 16 hours and phloretin (100 μM) before measure distance of migration by microscopy with 4× of magnification. (A) LX-2 cells were treated with succinate (1,600 μM) and phloretin (100 μM) for 16 hours. (B) LX-2 cells were treated with succinate (1,600 μM) and phloretin (100 μM) for 24 hours and measure collagen gel area. (C) Western blot analysis of tissue inhibitor of metalloproteases 1 (TIMP-1), phosphorylation myosin light chain 2 (p-MLC2), and MLC2 were detected using specific antibodies. Analysis of densitometry was performed and present data as the mean mean±standard error values of three independent experiments. GAPDH, glyceraldehyde-3-phosphate dehydrogenase. Statistical significance: aP<0.05, bP<0.01, and cP<0.001 vs. the control group; dP<0.05, eP<0.01, and fP<0.001 vs. succinate.
|
Phloretin reduced succinate-induced aerobic glycolysis in activated HSCs
In Fig. 3A, it was observed that the treatment of LX-2 cells with succinate resulted in an increase in mRNA expression of GLUT-1 and LDHA. However, co-treatment with phloretin showed that only phloretin at 100 μM inhibited the increased mRNA expression of GLUT-1 and LDHA proteins induced by succinate treatment. Phloretin at 100 μM inhibited the increased protein expression of GLUT1, HK II, and LDHA (Fig. 3B). In addition, treatment of LX-2 cells with succinate led to a downregulation of p-AMPK, but this effect was reduced in cells co-treated with phloretin (Fig. 3C). The ECAR data indicated that treatment with 1,600 μM succinate did not significantly increase glycolysis but treatment with phloretin reduced glycolysis in LX-2 cells (data was not shown). To validate these results, the experiment was repeated using succinate at a concentration of 400 μM, which induced glycolysis in LX-2 cells, while phloretin co-treatment reduced this effect, as indicated by the ECAR data (Fig. 3D).
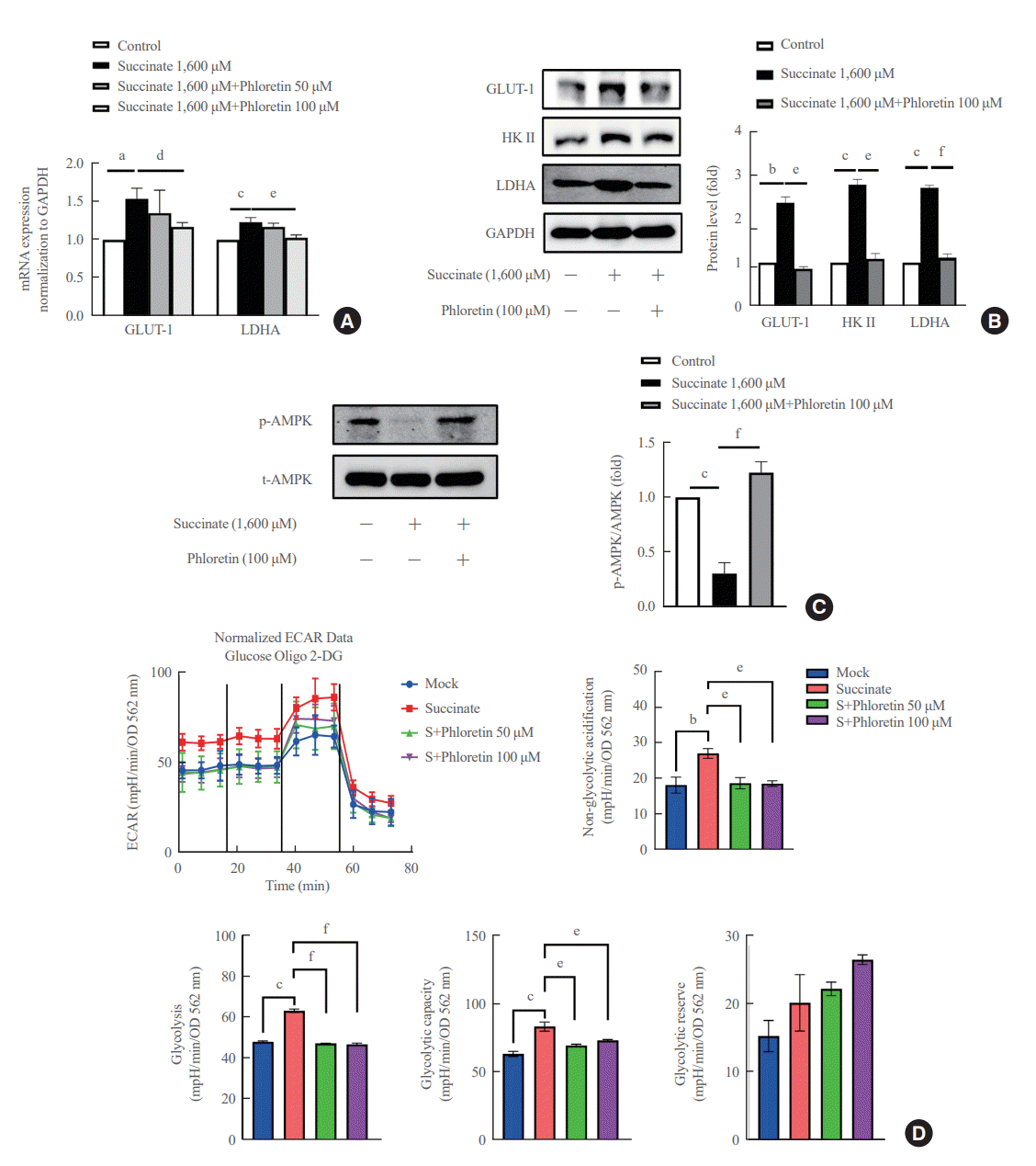 | Fig. 3.Phloretin reduced succinate-induced aerobic glycolysis in activated hepatic stellate cells (HSCs). LX-2 cells were treated with succinate (1,600 μM) and phloretin (50 or 100 μM) for 24 hours. (A) Real-time polymerase chain reaction to check mRNA expression of glucose transporter 1 (GLUT-1) and lactate dehydrogenase (LDH). (B) Western blot analysis of GLUT-1, hexokinase II (HK II), and lactate dehydrogenase A (LDHA) were detected using specific antibodies. (C) Western blot analysis of phosphorylated adenosine monophosphate protein kinase α (p-AMPKα) and AMPKα were detected using specific antibodies. (D) Seahorse analysis of extracellular acidification rate (ECAR), and glycolysis, glycolytic capacity and glycolytic reserve with succinate and phloretin (50 or 100 μM). Analysis of densitometry was performed and present data as the mean±standard error values of three independent experiments. GAPDH, glyceraldehyde-3-phosphate dehydrogenase; OD, optical density; 2-DG, 2-deoxy-D-glucose. Statistical significance: aP<0.05, bP<0.01, and cP<0.001 vs. the control group; dP<0.05, eP<0.01, and fP<0.001 vs. succinate.
|
Phloretin administration improved liver fibrosis induced by a sodium succinate diet in mice
Mice were fed a sodium succinate diet demonstrated an increase in serum ALT and hepatic triglyceride levels, which was reduced in animals receiving intraperitoneal injections of phloretin (Fig. 4A). In addition, mice treated with phloretin showed improved hepatic steatosis, as assessed by H&E staining, and hepatic fibrosis, as assessed by Masson’s trichrome staining, when compared to mice fed a sodium succinate diet without treatment (Fig. 4B). Fig. 4C also revealed that phloretin administration reduced the expression of α-SMA and collagen type 1, which had increased after 16 weeks of sodium succinate administration. These findings suggest that phloretin administration may protect against liver damage and inhibit the development of liver fibrosis in this animal model of sodium succinate-induced liver fibrosis.
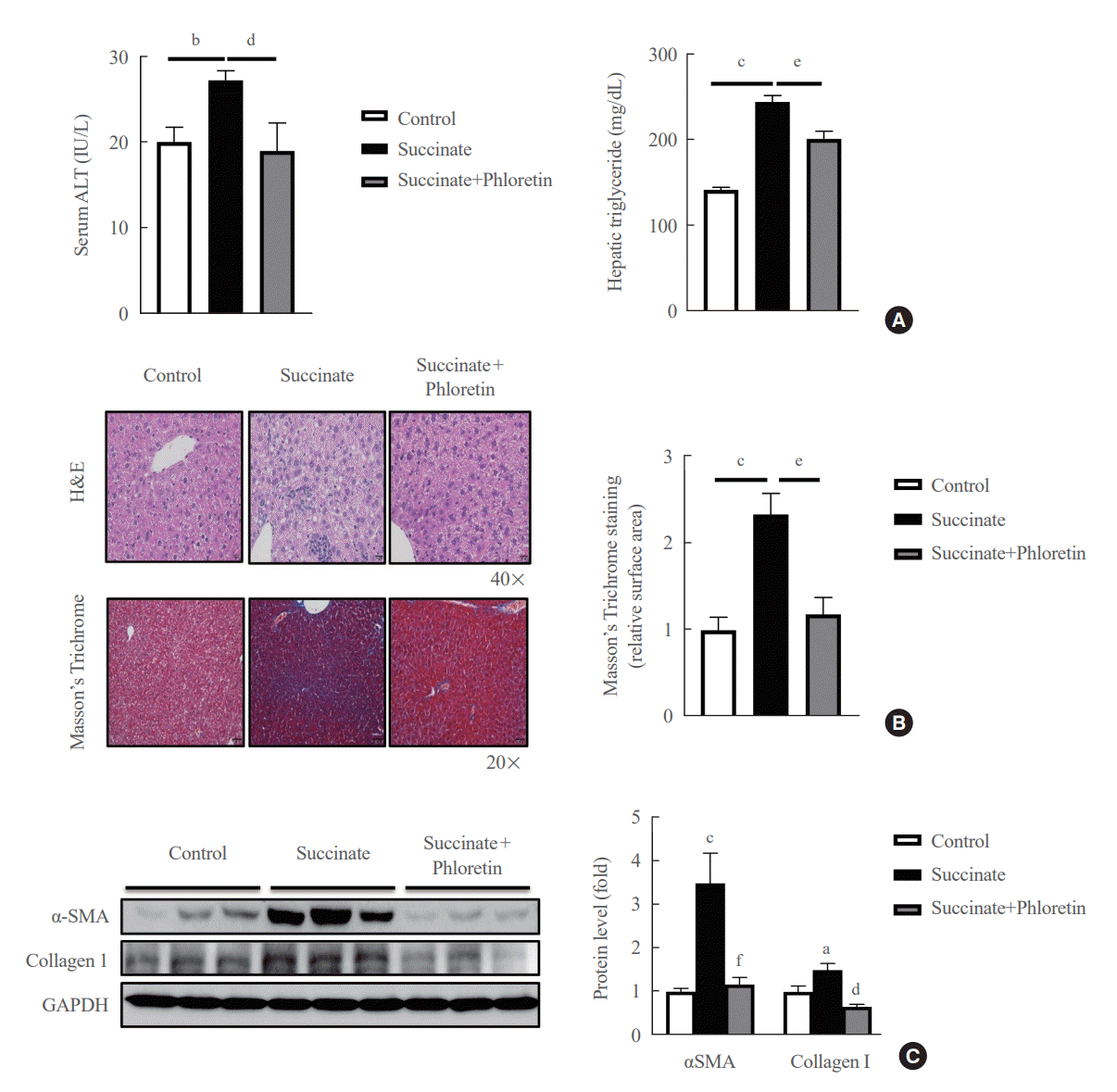 | Fig. 4.Phloretin administration improved liver fibrosis induced by a sodium succinate diet in mice. (A) Serum alanine transaminase (ALT) levels and hepatic triglyceride levels. (B) Hematoxylin and eosin (H&E) stain and Masson’s trichrome stain show the effect of phloretin intraperitoneal (10 mg/kg every other day) on sodium succinate 2% containing diet-induced liver fibrosis in mice. H&E staining 40×, Masson’s Trichrome 20×. (C) Liver from control group, sodium succinate 2% containing diet-feeding mice, and intraperitoneal phloretin group (10 mg/kg every other day) were used to analyze expression of α-smooth muscle actin (α-SMA) by Western blot. Analysis of densitometry was performed and present data as the mean±standard error values of three independent experiments. GAPDH, glyceraldehyde-3-phosphate dehydrogenase. Statistical significance: aP<0.05, bP<0.01, and cP<0.001 vs. the control group; dP<0.05, eP<0.01, and fP<0.001 vs. the succinate group.
|
Administration of phloretin decreased expression of glycolytic markers in the livers of mice with sodium succinate diet-induced liver fibrosis
We checked the product of glycolysis, lactate, in serum and liver tissue. The results in Fig. 5A demonstrated that phloretin reduced the lactate in serum and liver. Results of Western blots of the livers of mice fed a sodium succinate diet for 16 weeks showed increased levels of GLUT-1, and LDHA compared to control mice fed normal chow; this upregulation in protein levels of GLUT-1, LDHA was attenuated by administration of phloretin for 16 weeks (Fig. 5B). These results suggest that phloretin administration might inhibit the glycolytic pathway associated with the development of liver fibrosis.
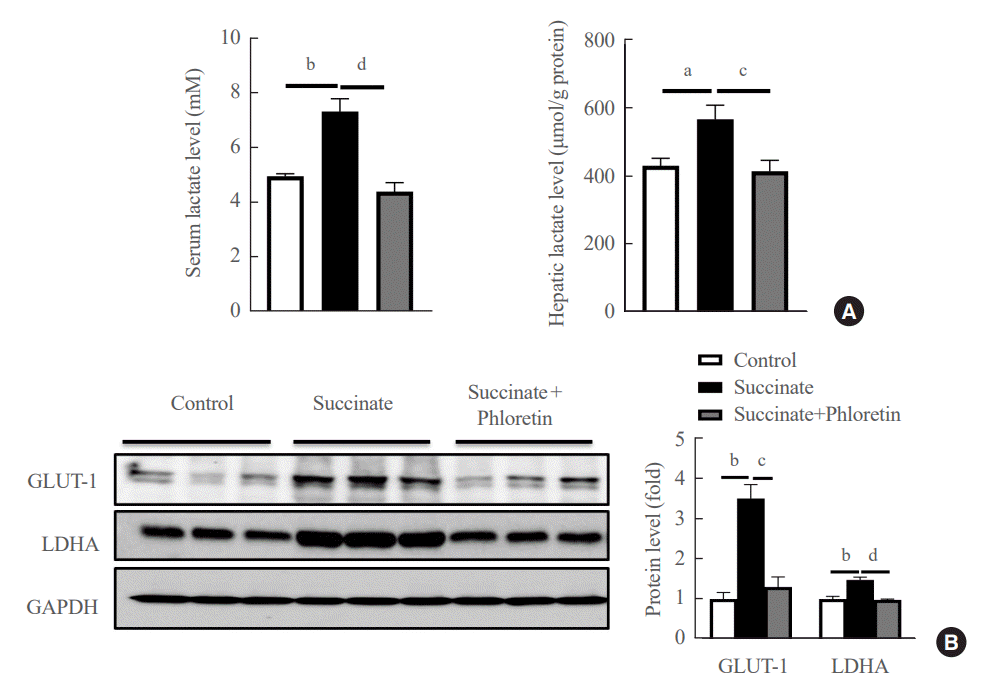 | Fig. 5.Administration of phloretin decreased glycolytic markers in the livers of mice with sodium succinate diet-induced liver fibrosis. (A) Hepatic lactate levels and serum lactate levels. (B) Liver from control group, sodium succinate 2% containing diet-feeding mice, and intraperitoneal phloretin group (10 mg/kg every other day) were used to analyze expression of glucose transporter 1 (GLUT-1), lactate dehydrogenase A (LDHA) by Western blot. Analysis of densitometry was performed and present data as the mean±standard error values of three independent experiments. GAPDH, glyceraldehyde-3-phosphate dehydrogenase. Statistical significance: aP<0.05, bP<0.01 vs. the control group; cP<0.05, dP<0.01 vs. the succinate group.
|
Go to :
DISCUSSION
In this study, we tested whether phloretin had therapeutic potential in treating hepatic fibrosis in a mouse model of sodium succinate diet-induced model of liver fibrosis. Furthermore, we also aimed to elucidate the role of aerobic glycolysis in the activation of HSCs and to discover the molecular mechanisms through which phloretin can reduce the activation of HSCs, the main source of the increased production of ECM components in liver fibrosis.
Liver fibrosis is a wound healing process to chronic liver injury, and activation of HSCs plays an important role in the production of the ECM during liver fibrosis. When quiescent HSCs are activated, HSCs exhibit increased proliferation, migration, and contraction [2]. The recent studies have shown that succinate, a TCA cycle intermediate, plays a vital role as a signaling molecule in both ischemic liver damage and chronic liver diseases [26]. One recent study demonstrated that adding succinate to drinking water can regulate the activation of thermogenic processes in adipose tissue [4]. In our previous study, we showed that succinate administration increased the expression of fibrogenic markers, upregulated inflammatory cytokine expression, and enhanced the proliferation and migration of LX-2 cells, and that administration of intraperitoneal succinate in mice resulted in increased α-SMA and inflammatory cytokine protein expression without inducing any obvious morphological changes [10-12]. In particular, a diet containing 2% sodium succinate resulted in increased steatosis and morphological changes associated with liver fibrosis, along with the accumulation of fibrogenic and glycolytic markers in the livers of mice. Furthermore, mice fed a sodium succinate diet exhibited increased levels of serum ALT and hepatic triglycerides, consistent with the changes observed in chronic liver damage. These results emphasize the vital role of succinate as an inducer of liver fibrosis.
At a molecular level, the current understanding of the role of HSCs suggests that their activation requires rapid energy reprogramming similar to the Warburg effect that occurs in tumor cells, and the survival of HSCs significantly depends on aerobic glycolysis [15]. This metabolic switch optimizes the consumption of glucose in HSCs and provides support for fibrogenic trans-differentiation [15]. Some recent studies have shown that the aerobic glycolytic activity regulating the activation of HSCs is modulated by the activation of AMPK [27], and inhibition of glycolysis can reduce the contractility of HSCs [28]. In the present study, we found that enhanced activation of aerobic glycolysis occurs during activation of HSCs. These results again verify the necessity of aerobic glycolysis in modulating the activation and function of HSCs, and the progression of liver fibrosis.
On the other hand, the latest study on phloretin, a well-known antioxidant compound, revealed that phloretin could mitigate obesity, maintain metabolic homeostasis, block infiltration of macrophages into white adipocyte tissue, and prevent diet-induced insulin resistance and hyperinsulinemia [21]. In this study, we have shown that intraperitoneal injection of phloretin improves hepatic steatosis and decreased hepatic fibrosis compared to untreated sodium succinate diet-induced mice. In the in vitro experiments, treatment with phloretin also inhibited the expression of α-SMA, collagen type 1, TIMP-1 in succinate-induced activation of HSCs. Furthermore, phloretin demonstrated to inhibit the proliferation, migration, and contraction of LX-2 cells induced by succinate during activation of HSCs.
Finally, we found that phloretin inhibited the upregulation of aerobic glycolysis that occurs during activation of HSCs. In particular, treatment with phloretin decreased the expression levels of glycolytic markers such as GLUT-1, HK II, LDHA, and decreased lactate production in LX-2 cells treated with succinate. In sodium succinate diet-fed mice model, administration of phloretin inhibited the increase of glycolytic proteins in the liver and reduced concentrations of serum and hepatic lactate. These findings suggest that phloretin might ameliorate the activation of HSCs and attenuate the development of liver fibrosis through the regulation of glycolytic processes. However, currently, the link between succinate signaling and glycolysis pathway was not well clarified. There is just one publication showed that stimulator of interferon genes (STING), a cytosolic sensor that regulates innate immune responses, increases the intracellular succinate concentration and succinate induces HIF-1α stabilization leading to increased glycolysis in macrophages with Brucella abortus infection [29].
In conclusion, this study demonstrated that administration of phloretin reduced expression the fibrogenic markers, inhibited the increased proliferation, migration, and contractility observed during HSC activation, and improved hepatic steatosis and fibrosis in a mouse model of sodium succinate diet-induced fibrosis. Furthermore, we found that aerobic glycolysis plays an essential role during the activation of HSCs and the progression of liver fibrosis in mice. The development of drug delivery systems using phloretin and the inhibition of aerobic glycolysis to reduce the activation of HSCs to further mitigate liver fibrosis remain promising avenues for further investigation.
Go to :
 XML Download
XML Download