

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Idiopathic pulmonary fibrosis (IPF) is a progressive chronic disease characterized by excessive fibrotic scarring of the lungs leading to irreversible decline in lung function.1
IPF pathogenesis is characterized by the progressive fibrotic change in the lung parenchyma.2 The process is thought to begin at subpleural area and progresses gradually into the lung parenchymal region.3 Considering the anatomical location, it would be a reasonable assumption that the visceral or parietal pleura plays an important role in the progression of lung fibrosis. However, the pathogenesis associated with fibrotic progression of the IPF has not been well studied.4
Alveolar injury in IPF promotes migration, activation, and proliferation of fibroblasts and myofibroblasts which produce collagen in the active regions of fibrotic change, also known as the fibroblastic foci.5 Fibroblastic foci are a characteristic histopathological feature of IPF/usual interstitial pneumonia (UIP).6 Honeycomb formation in IPF could be a result of airway remodelling after interstitial fibrotic scarring.5 The end result of these processes is a complex reticulum that is highly interconnected and extends from pleura into the underlying lung parenchyma.3
Cryptogenic organizing pneumonia (COP) is believed to be a consequence of alveolar injury and is characterized by the formation of organized fibrous tufts of granulation tissue obstructing the alveolar spaces and bronchioles.7 In contrast to IPF, these fibrotic lesions can be reversible.8
To date, studies evaluating pleural mesothelial cell properties in samples obtained from IPF or COP patients are rare.3
We presumed that pleural mesothelial cells play an important role in fibrosis activity9 and we aimed to determine whether pleural mesothelial cell markers expression reflects their profibrotic properties in IPF compared with COP.
METHODS
Patient selection
The inclusion period of this study was from August 1, 2002, to December 31, 2011, and the study was conducted at the Department of Internal Medicine, Keimyung University Dongsan Hospital, in Daegu, South Korea.
The patients underwent video thoracoscopic or open thoracic lung biopsy. The biopsy was mainly performed on the lung parenchymal tissue including subpleural lesions on the affected sides. As for the biopsy site, the area suspected of having an active lesion was selected on chest computed tomography (CT).
We used open lung biopsy or video-assisted thoracoscopic to evaluate interstitial lung disease (ILD). Forty-eight patients with ILD were enrolled in this study. Among them, 11 cases of connective tissue disease, 4 cases of nonspecific interstitial pneumonia, 2 cases of hypersensitivity pneumonitis, 1 case of desquamative interstitial pneumonia, 1 case of lymphocytic interstitial pneumonitis were excluded from the study. In the study were included 20 cases of IPF and 9 cases of COP of unknown etiology diagnosed based on pathologic and clinical features. There was no patient with a significant level of antinuclear antibody or rheumatoid factor titer in COP.
A biopsy was performed on patients who needed to be differentiated from COP on chest CT, before starting systemic corticosteroids in patients with COP. After the biopsy, oral prednisone 0.5 mg/kg was prescribed to 9 COP patients. Antifibrotic agents was not prescribed both IPF and COP.
Forced vital capacity (FVC) and diffusing capacity of the lungs for carbon monoxide (DLCO) were measured before lung biopsy as a baseline. A second lung function test was performed after an average of 3.7 months in patients with COP and 4.9 months in patients with IPF.
Lung immunohistochemistry
Paraffin sections were cut to 5 μm of thickness, mounted on silane-coated glass slides, and stored for 1 hours at 60°C. The slides were deparaffinized thrice with xylene for 5 minutes each, and rehydrated with graded alcohols (100%, 95%, 70%, and 50%) for 5 minutes, respectively. After washing with 0.01 M phosphate-buffered saline (PBS) for 5 min, the sections were digested with proteinase K (20 μg/mL) at room temperature for 20 minutes, and then washed twice with distilled water for 2 minutes each time. Endogenous peroxidase activity was blocked with 3% hydrogen peroxide (H2O2) in PBS for 5 minutes, and the slides were rinsed twice with PBS for 5 minutes. Sections for the positive control was treated with 3% H2O2 and then washed twice with PBS. For negative controls, the sections were covered with reaction buffer only and incubated under the same conditions. Sections were then incubated for 1.5 hours with monoclonal antibodies against leucine rich repeat neuronal 4 (LRRN4), uroplakin 3B (UPK3B), CC-chemokine ligand 18 (CCL18), and laminin-5 and after washing incubated again with biotinylated secondary antibodies. The antibody reactions were visualized using diaminobenzidine as chromogen (DAKO, Carpinteria, CA, USA). For microscopic observation, the sections were lightly counterstained with hematoxylin for 1 minute.
Semi-quantitative analysis of immunohistochemistry
Immunohistochemical (IHC) staining was performed using pleural mesothelial cell markers, LRRN4 (Sigma-Aldrich, St. Louis, MO, USA) in 1:25 dilution and mesothelial cell localization,10 UPK3B (Novus, Centennial, CO, USA) in 1:500 dilution and mesothelial cell localization,10 also markers known to be associated with IPF pathogenesis, CCL18 (Sigma-Aldrich) in 1:25 dilution and role in induction of collagen synthesis by fibroblasts11 and laminin-5 (Abcam, Cambridge, UK) in 1:500 dilution and role in regenerating epithelial cells.12
Sections were reviewed by the lead investigator alongside an independent pathologist and scored by examining the expression of the IHC markers at sites of smooth muscle bundles, macrophages, alveolar lining cells, endothelial cells, fibroblastic foci, alveolar lining cells, bronchioles, honeycombing, bronchiolar metaplasia and fibrotic lesion in COP. A total of 100 pleural mesothelial cells were counted and the number of cells expressing each marker were recorded in each area. Semi-quantitative analysis was used to compare the groups using a modified Allred scoring system.13
The staining score 0 indicated positively stained cells at 0%, a staining score of 1, indicated positively stained cells to be < 25%, a staining score of 2, indicated positively stained cells to be in the range of ≥ 25% to < 50%, and a staining score of 3, indicated positively stained cells to be ≥ 50%.
Statistical analysis
The Mann Whitney U test was used to compare markers expression between IPF and COP samples. The results are presented as the mean ± standard deviation (SD) score for each marker. Staining group values were expressed as means ± SD. Analysis of variance FVC % predicted value and for comparison of the different groups was used with significance set at P < 0.05. A significant analysis of variance was followed by a Scheffe test for multiple comparisons between groups, again with a P < 0.05. A paired t-test was used to compare the baseline and follow-up FVC and DLCO. Differences were considered significant if the P value was < 0.05. Statistical analyses were performed using IBM SPSS Statistics for Windows (IBM SPSS version 21.0.; IBM Corp., Armonk, NY, USA).
Ethics statement
The current study was approved by the Institutional Review Board (IRB) of Keimyung University, Dongsan Hospital (2010-02-004). The IRB waived the requirement for informed consent due to retrospective nature. All methods were performed in accordance with the relevant guidelines and regulations.
RESULTS
Characteristics at baseline
The average age of the IPF and COP patients was 65 years, and in the case of IPF, the patients were predominantly male. The mean FVC values at baseline were similar between IPF and COP groups (Table 1). The biopsy specimens contained pleura and sufficient lung parenchyma evaluating broncho-vascular bundle in both diseases (Fig. 1).
Table 1
Baseline patient characteristics (N = 29)
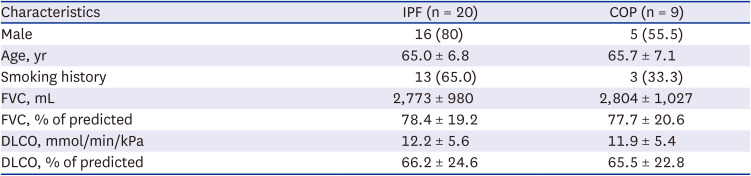
Fig. 1
Comparison of microscopic findings of representative IPF and COP. (A, B) IPF: Patchy fibrosis and preserved alveolar structures are seen in the lung parenchyma (A). In enlarged view, irregular fibrotic thickening was observed, and fibroblastic foci (arrows) could be observed (B). (C, D) COP: Fibrous tufts (arrows) filling the alveolar cavity can be observed diffusely.
IPF = idiopathic pulmonary fibrosis, COP = cryptogenic organizing pneumonia.
IHC stain
LRRN4 expression
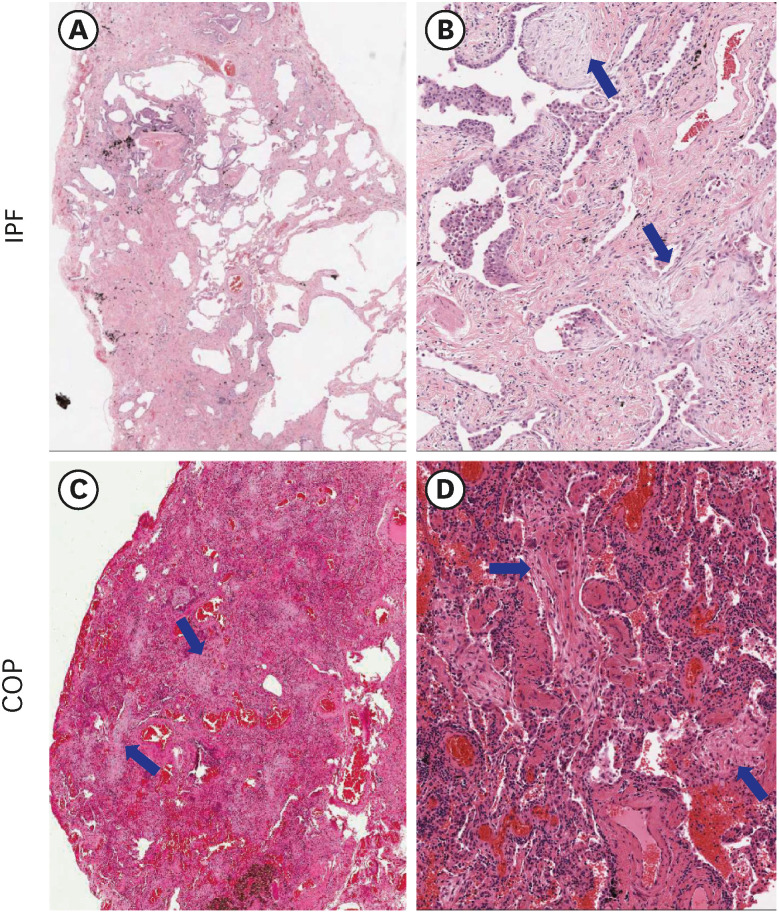
In IPF group, significantly higher expression was observed in the epithelial cells covering fibroblastic foci, honeycomb and some endothelial cells compared to COP group. In COP group, expression was negative or weak in the epithelial cells surrounding the fibrous tufts (Table 2, Fig. 2). The positive expression of LRRN4 in the mesothelial lining cells, was a clear indication that this protein is a mesothelial cell marker. LRRN4 seems to reflect an active fibrotic process, and its intense expression enhances the number of fibroblastic foci (Table 2).
Table 2
Semi-quantitative analysis of targeted marker expression IPF and COP
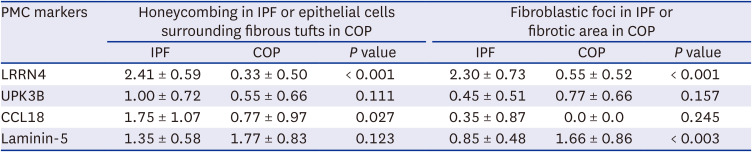
Fig. 2
Comparison of immunohistochemical staining results for LRRN 4, UPK3B, CCL18, and laminin-5 in between IPF and COP samples. LRRN4: In IPF, positive expression in the epithelial cells along the honeycombing (A) and cells covering the fibroblastic foci (B, arrow). In COP, no expression in the epithelial cells surrounding fibrous tufts (C). UPK3B: In IPF, mild expression in the epithelial cells covering honeycombings (D) and positive expression in the pleural mesothelial cells (E, arrows). In COP, weak expression in the fibrous tufts (F). CCL18: In IPF, positive expression in the smooth muscle bundles (G, arrows), positive expression in the alveolar epithelial cells (arrow), and alveolar macrophages (arrow heads) in fibrotic lesion (H). In COP, no expression in the fibrotic area (I). Laminin-5: In IPF, focal expression in honeycombing (J, arrows) and in the epithelial cells covering fibroblastic foci (K, arrows). In COP, diffuse expression in the epithelial cells surrounding the fibrous tufts (L). LRRN4 and UPK3B were clearly stained in the visceral pleura both IPF and COP samples. All markers were no or scanty staining in normal epithelium. Original magnification: A-C and F-L (×100); D and E (×40).
LRRN4 = leucine rich repeat neuronal 4, UPK3B = uroplakin 3B, CCL18 = CC-chemokine ligand 18, IPF = idiopathic pulmonary fibrosis, COP = cryptogenic organizing pneumonia.

UPK3B expression
In IPF, UPK3B showed mild expression in the epithelial cells covering honeycombing and positive expression in the pleural mesothelial cells. In COP, weak expression was observed in fibrotic areas (Fig. 2). We did not observe any significant difference in UPK3B expression between the IPF and COP groups (Table 2).
CCL18 expression
In IPF, CCL18 was expressed in the bronchiolar metaplasia, fibroblastic foci, epithelial cells along the honeycombing, but CCL18 expression was absent in the fibrotic area of the COP (Table 2). In both IPF and COP, the fibrotic scarring was frequently associated with the smooth muscle proliferation and higher CCL18 expression (Fig. 2).
Laminin-5 expression
In IPF, laminin-5 was expressed in the epithelial cells covering fibroblastic foci, and we observed focal and weak staining in honeycombing. Fibrotic scar lesion was not stained, but bronchiolar epithelial cells were positively stained, and re-epithelialized alveolar cells were weakly or focally stained. In COP, laminin-5 was expressed in the epithelial cells surrounding the fibrous tufts (Fig. 2). There was no significant difference in laminin-5 expression between the COP and IPF samples (Table 2).
FVC and DLCO in IPF and COP
Nine COP patients were treated with 0.5 mg/kg prednisolone for 1–3 months and showed improvement in percent predicted FVC (88.4 ± 18.7, P < 0.01) and DLCO (72.8 ± 21.1, P < 0.01). There was no case of lung function decline after corticosteroid therapy in patients with COP. In IPF, there was no significant change in percent predicted FVC (77.8 ± 19.5) and DLCO (63.4 ± 23.3) values between baseline and follow-up.
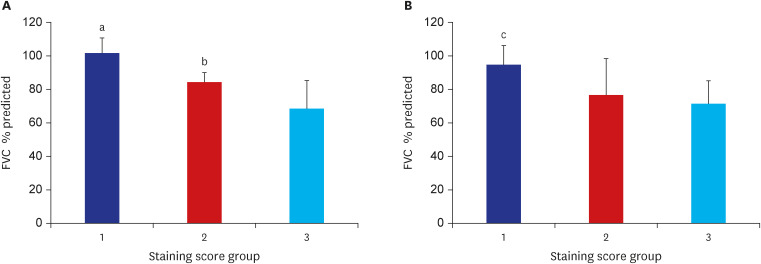
High-intensity staining (staining score 3 group) of LRRN4 in epithelial cells along the honeycombing showed significantly low FVC % predicted compared with weak staining in staining scores 1 and 2 groups (Fig. 3A). LRRN4 staining in fibrotic foci showed group 3 (staining score 3) had a significantly lower FVC % predicted value than group 1 (staining score 1) (Fig. 3B).
Fig. 3
Staining score group and FVC % predicted. (A) The staining score of LRRN4 in the epithelial cells along the honeycombing and FVC % predicted. Twenty patients with idiopathic pulmonary fibrosis were divided into 3 groups based on LRRN4 staining score in the epithelium. Staining scores 1, 2, and 3 represent 3, 6, and 11 patients, respectively. (B) The staining score of LRRN4 in fibrotic foci and FVC % predicted. Twenty patients with idiopathic pulmonary fibrosis were divided into 3 groups based on LRRN4 staining score in the fibrotic foci. Staining scores 1, 2, and 3 represent 4, 6, and 10 patients, respectively.
FVC = forced vital capacity, LRRN4 = leucine rich repeat neuronal 4.
There was a significant difference between LRRN4 staining group 1 and group 2: aP = 0.016 vs. group 3, bP = 0.035 vs. group 3. There was a significant difference between LRRN4 staining group 1 vs. group 3 cP = 0.026.
DISCUSSION
In this study, we found that LRRN4, which is considered a pleural mesothelial cell marker, is expressed in epithelial cells along the honeycombing and fibroblastic foci, which are the most important areas in IPF pathogenesis. On the other hand, LRRN4 was not expressed in COP. We suggest that pleural mesothelial cell properties may be key players in fibrosis activity in IPF. Higher expression of LRRN4 may be associated with a decline in FVC. This gives us a clue that LRRN4 might be a predictive factor for the severity of fibrosis, but it needs to be further studied.
The reason for comparing the differences in the mesothelial cell markers expression in IPF and COP is that, among the idiopathic interstitial pneumonias, IPF and COP would have different pathogenesis considering COP could be reversible.
A balance between epithelium and mesenchymal cells is essential to maintain homeostasis in adult tissues.14 Mesenchymal cells differentiate into various cells such as bronchial smooth muscle cells, vascular smooth muscle cells, and fibroblasts which can promote mesothelial-mesenchymal transition of pleural mesothelial cell and play a central role in repairing airways, alveoli, and blood vessels.15 We suggest that expression of pleural mesothelial cell markers are crucial mediators of fibrosis.
In IPF, IHC result showed positive expression of LRRN4 in the epithelial cells along the honeycombing, fibroblastic foci and some endothelial cell. In COP, it showed weak expression in epithelial cells surrounding fibrous tufts (Table 2, Fig. 2). Higher LRRN4 expression reflects reduced lung volume and increased fibrotic activity in IPF. A follow up pulmonary function test showed that FVC was reversible in COP, but not in IPF. LRRN4 positive expression in IPF, and negative expression in COP, may indicate its role in IPF pathogenesis. Further studies are needed to provide more evidence regarding the profibrotic properties of pleural mesothelial cell and the relationship between LRRN4 and fibrotic activity.
We observed UPK3B expression in the epithelial cells covering honeycombing, in pleural mesothelial cells and weak expression in fibrotic areas in COP. However, there was no significant difference in UPK3B expression was detected between IPF and COP groups.
In the case of CCL18, there are reports showing its relationship to lung fibrosis and the prognosis of IPF.161718 Few studies have been on the morphological expression of CCL18 in IPF. In this study, CCL18 expression in alveolar macrophages, epithelial cells, and smooth muscle bundles suggests that CCL18 activity is not limited to the epithelium, but it is also expressed in the interstitium as it was stated in other studies.1920 This suggests that CCL18 may also play a role in IPF pathogenesis.16
Thus, it can be inferred that the involvement of pleural mesothelial cells plays a key role in the irreversibility of lung fibrosis.21
On thin-section high-resolution CT, predilection site for IPF is subpleural area, in contrast to COP, which involves the intraalveolar area especially along the bronchovascular bundles.2223 In our study LRRN4 was expressed in structures close to the visceral pleural in IPF but not in COP group. Although we did not study LRRN4 expression during follow-up, LRRN4 was found to be negatively correlated with lung volume in IPF patients at baseline. Fibrosis in IPF is tightly connected with the pleura.3 Therefore, we suggest that fibrotic activity closer to the visceral pleura may be more irreversible.3
There are several limitations to this study. First, as mesothelial cell markers, mesothelin, cytokeratin 5/6, WT1 and calretinin are also available, but in this study, the association between these markers and fibrosis did not evaluate. Second, a comparison to normal lung tissues would be required to appropriately understand the general unperturbed localization of mesothelial cells compared to these diseases’ tissues. Third, this study is a descriptive study and has limitations in proving the causal relationship between PMC and fibrosis. Fourth, COP may cause progressive fibrosis. However, COP can manifest as acute fibrotic organizing pneumonia (AFOP) and lead to progressive fibrosis. We did not include AFOP for these COP subgroups in our study.
In conclusion, LRRN4 expression patterns in IPF are distinct from those in COP. Our findings suggest that pleural mesothelial cells profibrotic property may be an important player in IPF pathogenesis and could be a clue in the irreversibility of fibrosis in IPF. CCL-18 expression in human tissue samples provides new insights into IPF pathogenesis.
 XML Download
XML Download