

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Primary cilia are antenna-like organelles projecting from the cellular membrane. These tubulin-based structures are highly conserved across the species and found on almost every cell in the human body [1]. Cilia in the form of flagella on swimming protozoa were discovered in the 1670s by the great Dutch microscopist, Van Leeuwenhoek, father of microbiology [2]. It would take another 200 years for the discovery of primary cilia on mammalian cells in 1898 [3]. Unlike motile cilia whose role in human genetic diseases was established early on [4], the function of primary cilia had not been elucidated until these past two decades [5]. Since then, exploding interest in cilia have brought this once forgotten organelle to the forefront of cell biology and human disease research. Through recent advances in proteomics, genetics, and microscopy, we are starting to piece together aspects of primary cilia function through the characterization of receptors, ion channels and signaling molecules that are uniquely enriched and interactive in primary cilia [6-9]. At this point, primary cilia are recognized as important signaling hubs for cellular development, differentiation, and function, rather than vestigial organelles [8,10-12].
As cilia are highly conserved and present on most cells of vertebrate organisms, they play a central role in normal development and also in disease. In humans, inherited motile and primary cilia disorders are collectively termed ciliopathies and involve nearly all major organs [1]. Most early studies on cilia function had been limited to motile cilia [4,13], namely the complex motile ciliopathy syndrome known as primary ciliary dyskinesia which includes Kartagener’s syndrome (also known as immotile cilia syndrome) characterized by a triad of situs inversus, chronic sinusitis, and bronchiectasis resulting from cilia dysmotility [4]. The year 2000 saw the seminal discovery of hypomorphic mutation in Tg737, or intraflagellar transport 88 (IFT88), an essential gene for cilia assembly, where Tg737orpk mutant mice displayed the phenotype of autosomal recessive polycystic kidney disease [5] and prompted further investigation of the role of primary cilia in human disease. To date, multiple human genetic diseases have been linked to primary cilia dysfunction including autosomal dominant/recessive polycystic kidney disease, Bardet-Biedl syndrome (BBS), Alström syndrome, Meckel-Gruber syndrome, and Joubert syndrome [14-19]. The kidney, liver, brain, and retina are typically the organs most affected by primary ciliopathy and have been the traditional focus of ciliary studies [20-23]. Meanwhile, the association between cilia and energy metabolism found in BBS has sparked investigations into ciliary function in metabolic disorders such as obesity and diabetes [19]. In this review, we will summarize current knowledge about primary cilium structure, expression, and function in pancreatic islets, one of the regulatory centers of glucose and energy homeostasis in the body.
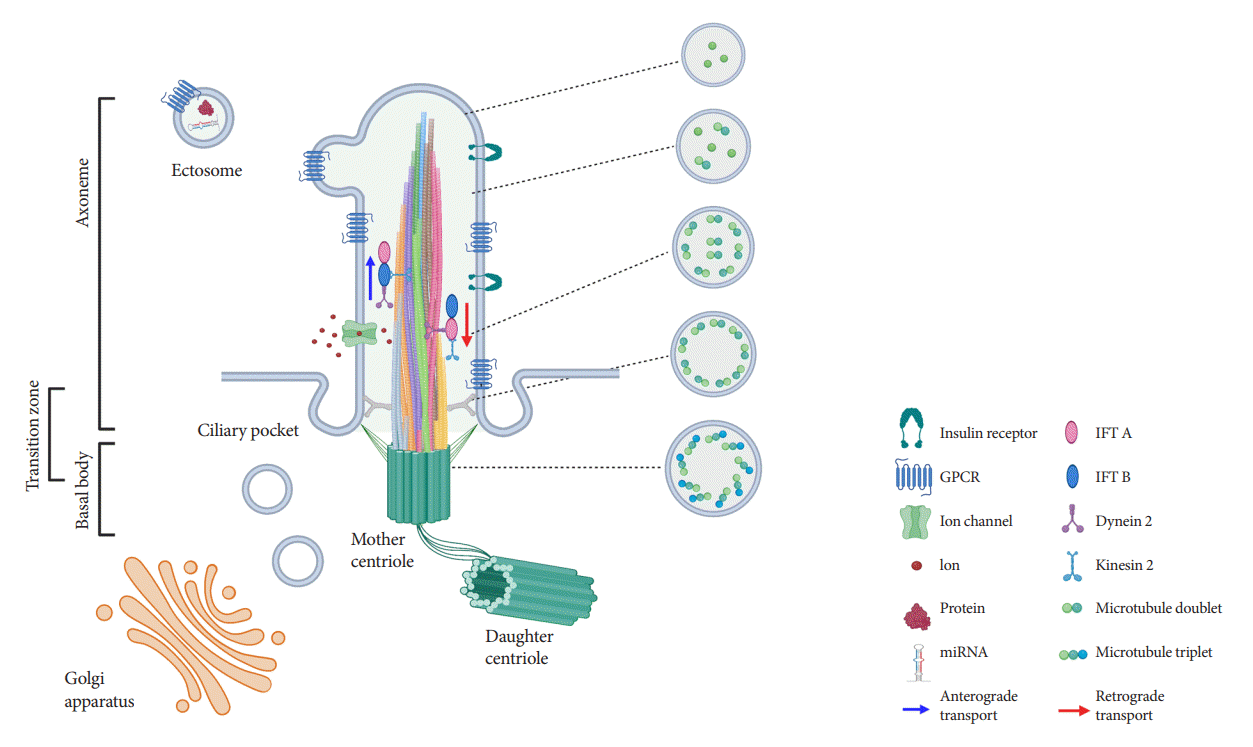
REVISITING THE STRUCTURE OF PRIMARY CILIA
Ultrastructural examination of primary cilia cross-sections by electron microscopy typically reveals a cylindrical axoneme composed of nine outer microtubule doublets with no central pairs, dubbed as the “9+0” configuration, in contrast to the motile cilia “9+2” paradigm where a central microtubule pair and associated structural motifs confer motility to the axoneme. However, recent findings by serial selection- or cryoelectron tomography have challenged this dichotomous classification, revealing that primary cilia have a dynamic structure that does not always conform to “9+0” expectations [24-26]. The axoneme of the primary cilia starts with “9+0” microtubules at the base, but not all microtubules have equal length, so they decrease in number as the axoneme grows distally (Fig. 1) [24,25]. In cultured kidney cells, primary cilia have been seen to adopt a “7+2” microtubule configuration immediately after leaving the basal body and become “3+0” toward the tip [24]. In addition to microtubule length and number, the arrangement of microtubules also changes from base to tip, reducing their pairing from triplet to doublet to singlet [24,25]. Along with these extra-pancreatic observations, several pioneering studies in pancreatic β-cell primary cilia have reported microtubule patterns including “8+2,” “7+2,” “3+0,” or “2+0” [27-29]. It appears that, while most primary cilia start as 9+0 at the base, they soon become something else, and the cross-sectional structure differs depending on where you look. Thus, these non-“9+0” arrangements, once regarded as atypical manifestations, may in fact reflect the normal evolution of primary cilia structure from base to tip.
The fact that most primary cilia diameter follows a gradual tapering from base to tip provides several clues that may explain the flexural bending properties, e.g., to extracellular flow and suggests greater complexity in the structure of primary cilia than previously assumed. Primary cilia are dynamic organelles that constantly undergo remodeling and whose functions depend on a busy intra-ciliary transport system [30,31]. Since at least half of the axoneme consists of microtubule singlets according to the recent findings [24,25], it raises questions about the current paradigm of a clear division between anterograde and retrograde IFT complexes on B- and A-tubules, and how bi-directional cargo trafficking might be accomplished on single microtubule filaments. Around 1,000 proteins have been identified in cilia-basal body complex, many of which are still of unknown function [32]. How these proteins are implicated in cilia assembly and maintenance, and how the intra-ciliary transport system functions in non-binary structures remain to be clarified. In addition to the evolving classification system based on cilia structure, there are also blurring distinctions of ciliary protein functions inside and outside the cilium. Growing evidence supports cilia function outside its membranebased structure through the release of extracellular vesicles (EVs) (Fig. 1), which serve to transmit long-range signals between cells [24,25,33-37]. EVs derived from primary cilia are heterogeneous and show signal-dependent ectocytosis [34,37]. The physiologic relevance of ciliary EVs in intercellular communication and function is entirely unstudied in pancreatic islets and therefore represents an opportunity for future investigation.
PRIMARY CILIA IN PANCREAS AND ISLETS
Ciliated cells in mouse and human pancreas
Cytoplasmic projections between the intercellular spaces were observed in β-cells of rabbits and guinea pigs in 1957 [38], and a year later these structures were identified as primary cilia in mouse β-cells, convincingly shown as a defined filamentous axoneme connected to the basal body and centrioles [39]. In early reports, islet cilia were only described in β-cells and not in other islet cells such as α-cells, ductal cells, or acinar cells. Subsequent studies have since demonstrated the presence of primary cilia in α-, δ-, and pancreatic polypeptide (PP) cells in mouse and rat pancreatic islets by electron and light microscopy (Table 1) [16,27-29,40-44]. In humans, primary cilia in the pancreas were first reported in β-cell tumor cells in 1964 [45]. So far, in human islets, only β- and α-cells have been documented to have primary cilia [42,45], whereas in the exocrine pancreas, primary cilia have been identified on ductal and centroacinar cells in both human and mouse [16,27,29,43]. RNA sequencing studies have demonstrated robust expression of cilia-related genes across cell types in mouse and human pancreas and islets, whose expressions are dynamically modulated by metabolic conditions such as hyperglycemia and diabetes mellitus [46-49]. These observations confirm near-ubiquitous expression of primary cilia in pancreatic tissues across species and implicate adaptability of cilia function in physiology and disease states.
Orientation of primary cilia in β-cells
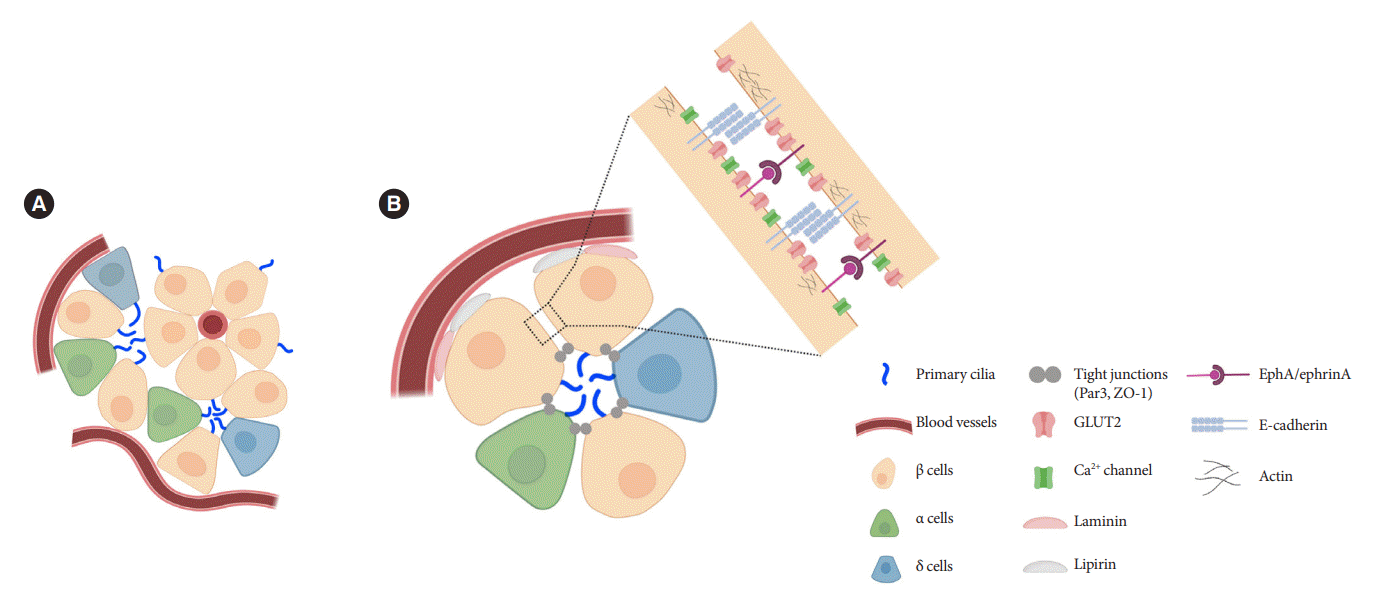
Islets are enclosed organoids containing endocrine cells arranged in a three-dimensional sphere, where cellular polarity and orientation are neither fixed nor fully understood. Emerging data suggest that rodent β-cells are polarized with regional specializations, such that there is lateral localization of glucose transporters and basal distribution of insulin granule [50-52]. The precise location of primary cilia on β-cells has been a challenge to define. Limited data by electron microscopy have shown conflicting results, with the cilium protruding either from the lateral surfaces of β-cells into the canalicular lumen or towards the vasculature [39,53]. It was unclear whether cilia orientation was conserved, or indeed mattered, until a finding in 2009 revealed that the pancreas-specific liver kinase B1 knockout mice exhibited altered β-cell polarity and ciliary orientation relative to blood vessels, showing for the first time that cilia orientation was regulatable in islet cells and may be linked to key β-cell functions such as cell size control and insulin secretion [54]. However, the issue remained that β-cell functional domains themselves were not well-characterized, thus there lacked reference points by which to define the spatial location of cilia and their polarity and orientation. Later studies contributed clarity to this issue by defining functional edge domains of the β-cell in mouse and human islets, showing consistent polarity determinants using three-dimensional confocal and serial electron microscopy [55-57]. The three distinct domains of pancreatic β-cells include the apical domain away from blood vessels, the lateral domain adjacent to neighboring β-cells, and the basal domain in contact with blood vessels (Fig. 2) [55]. In focused ion beam-scanning electron microscopy (FIB-SEM), β-cell primary cilia are found to project toward the extracellular apical lumen (Fig. 2) and along the apical tight junction (as marked by zonula occludens-1) while being away from the vasculature [55]. These findings together with earlier electron microscopy studies helped establish the current consensus view which is that islet cilia protrude into the canalicular lumen at intercellular junctions in the islet interior [53]. Compared to renal or vascular endothelial cells, in which the apical orientation of primary cilia is required for their role to detect flow or shear stress [58-60], the functional implication of apical-lateral positioned primary cilia in β-cells remains to be clarified. One potential explanation is that the apical orientation of primary cilia may enhance β-cell interaction with neighboring cells and potentiate autocrine and paracrine signaling [55]. Recently, we demonstrated that primary cilia not only have sensory but also motile function [49] which speculatively would better enable three-dimensional sampling of external cues. It would be of interest to examine ciliary motility as determined by its subcellular location and what varying degrees of freedom are afforded by its surrounding physical space.
Motility of primary cilia in pancreatic islets
Traditionally, cilia have been classified as motile and non-motile cilia according to their microtubule structure (“9+2” vs. “9+0”), as the central microtubule pair is thought to be essential for active movement. However, exceptions to this rule exist, as both non-“9+2” motile cilia and “9+2” immotile cilia have been described, thus the “9+2” structure appears neither necessary nor sufficient for ciliary motility. Nodal cilia in the developing embryo, for example, have a “9+0” axonemal structure and are famous for their rotational movement that drives morphogen flow, which serves in L–R axis determination [61- 65]. Meanwhile, the “9+2” kinocilia in auditory hair cells are non-motile and represent one of the heterogeneous cilia forms in the organ of Corti [66]. In any given tissue or organ system, it is likely that neither the “9+0” nor “9+2” form is absolute, and there may exist both inter-cilia as well as intra-cilia differences, since the microtubule structure of even a single primary cilium can change from base to tip [24,25]. Thus, primary cilia may require a more permissive classification, one that includes the possibility of motility in addition to their predominant sensory function.
While primary cilia have been reported to move passively, in response to extracellular fluid flow [58,67] or to tugging by intracellular actin forces [68], active motility by primary cilia had not been demonstrated until recently. Two studies in human pancreatic islets have reported that islet cells express motile cilia genes and protein complexes that were once thought to only exist in classic motile cilia [48,49]. These include the core axonemal dyneins dynein axonemal intermediate chain 1 (DNAI1), dynein axonemal heavy cahin 5 (DNAH5), dynein axonemal light chain 1 (DNAL1), as well as central pair-associated proteins sperm flagellar 2 (SPEF2) and kinesin family member 9 (KIF9), with their ciliary localization confirmed by light and electron microscopy in human islet cells [49]. These motile components mediate active movement of primary cilia via adenosine triphosphate hydrolysis, a key energy-generating reaction in living cells [49]. Functionally, β-cell cilia movement occurs in response to extracellular glucose and is coupled to glucose-stimulated events including Ca2+ influx and insulin secretion [49]. Cilia motility genes are dynamically expressed in islets, with enrichment in pancreatic endocrine cells compared to non-endocrine cells, and greater expression in diabetic than healthy islets [49]. Consistent with these findings, gene expression network analysis in human pancreatic islets showed that cilia-related genes in α- and β-cells, including motility genes, are upregulated in individuals with type 2 diabetes mellitus, and that modulation of these genes led to changes in glucoseinduced insulin secretion [48]. Taken together, these results reveal dynamic cilia expression in health and disease and implicate a new functionality of primary cilia that bridges the sensory and motile divide, as well as a regulatory role for cilia motility in β-cell insulin secretion. Future studies might elaborate how cilia motility works in orchestrating β-cell coupling within islets as well as in mediating heterotypic cell communications via paracrine or juxtracrine signaling.
Role of primary cilia in pancreas development
Primary cilia are dynamic organelles whose assembly and disassembly are linked to the cell cycle and differentiation status of the cell [69-71]. The role of primary cilia in pancreas development has been studied using genetic mouse models targeting ciliogenesis or cilia function (Table 2) [16,40-43,54,72-81]. The global Ift88 homozygous mutant mice present with absent or shortened pancreatic cilia, which in the pancreas exhibits as reduced pancreas mass with collagen deposition, progressive acinar cell loss, and ductal hyperplasia with pancreatic cyst formation [16]. These exocrine defects are consistent with phenotypes seen in knockout mouse models of ciliary protein Chibby1 or pancreas-specific kinesin family member 3A (Kif3a), an essential gene for cilia formation [43,72]. Proposed mechanisms for these pancreatic morphological changes include defective exocytosis of zymogen granules, intrapancreatic activation of digestive enzymes such as carboxypeptidase, and activation of transforming growth factor β and mitogen-activated protein kinase kinase/extracellular signal-regulating kinase pathways [41,43,72]. Similar pancreatic phenotype of the whole-body Ift88 mutant mice were also observed in mice harboring mutations in the polycystin 2 (Pkd2) and inversin genes, which mediate cilia function rather than cilia assembly [16]. These results indicate that both accurate assembly and function of primary cilia are required for proper pancreatic tissue organization. Of interest, induction of Kif3a loss-of-function at 4 weeks of age rather than prenatally failed to elicit a significant pancreatic phenotype, suggesting that there may be a critical period which requires normal cilia for pancreas development [72].
In contrast to in the exocrine pancreas, ciliary phenotypes in endocrine pancreas were less clear in the early studies, given conflicting results about their requirement in islet cell development and function [16,40-43,72]. The Chibby1- or pancreas-specific Kif3a-knockout mice exhibited grossly normal islet structure and apparently unperturbed endocrine cell differentiation, resulting in animals with normal glucose level or tolerance [43,72]. The hypomorphic Ift88 mutant mice showed comparable islet areas and β-cell maturity marker expression as in wild-type mice [16]. However, Ift88 hypomorph animals showed impaired glucose tolerance when challenged with high glucose, suggesting that primary cilia may be involved in the sensing or regulation of blood glucose levels [41]. Supporting this notion, whole-body deletion of regulatory factor X3 (Rfx3), a key ciliogenic gene, and β-cell specific deletion of Ift88 (β-cell specific cilia knockout [βCKO]) both pre- and post-natally produced reduced or absent cilia, which led to impaired glucose tolerance and decreased insulin secretion [40,42]. Both these cilia deletion models showed altered islet cell composition, suggesting cilia involvement in islet development. In Rfx3–/– mice, mRNA and/or protein expression of insulin, glucagon, and ghrelin decreased 10- to 150-fold while PP increased 15-fold [40]. β-Cell specific Ift88–/– mice showed reduced β-cell mass, insulin content and secretion, while δ-cell mass and somatostatin secretion were increased [42]. In another inducible β-cell Ift88–/– βICKO mice, β-cell mass was normal in the early period of cilia deletion but progressively decrease over time: normal 6 weeks after cilia deletion, to 10% decrease after 8 weeks, and over 80% decrease after 20 weeks [42,79]. These findings suggest that primary cilia play a requisite role in pancreatic endocrine cell differentiation as well as maintenance of islet cell mass. Consistently, cilia-dependent Gli/Hedgehog activation in mice causes β-cell dedifferentiation, providing a potential mechanism by which primary cilia regulate islet cell fate [82,83]. Further studies are needed to elucidate how primary cilia regulate the development of specific islet cell subsets and to identify ciliary signaling pathways linked to endocrine cell differentiation.
ROLE OF CILIA IN ISLET HORMONE RELEASE
Emerging consensus from both in vitro and in vivo studies show that primary cilium defects significantly impair β-cell function by reducing both insulin production and secretion [40,42,79,80]. In βCKO mice, deletion of β-cell Ift88 attenuated first-phase insulin secretion and delayed recovery to baseline secretion compared with wild type [42], indicating that that primary cilia regulate β-cell synchrony in their secretory response to glucose [42,84]. Similar results were reported in tamoxifen-inducible cilia knockout mice [42,79], demonstrating that the removal of cilia function in mature functional β-cells, rather than the developmental changes that result from cilia deletion, accounted for the β-cell phenotypes in these experimental models.
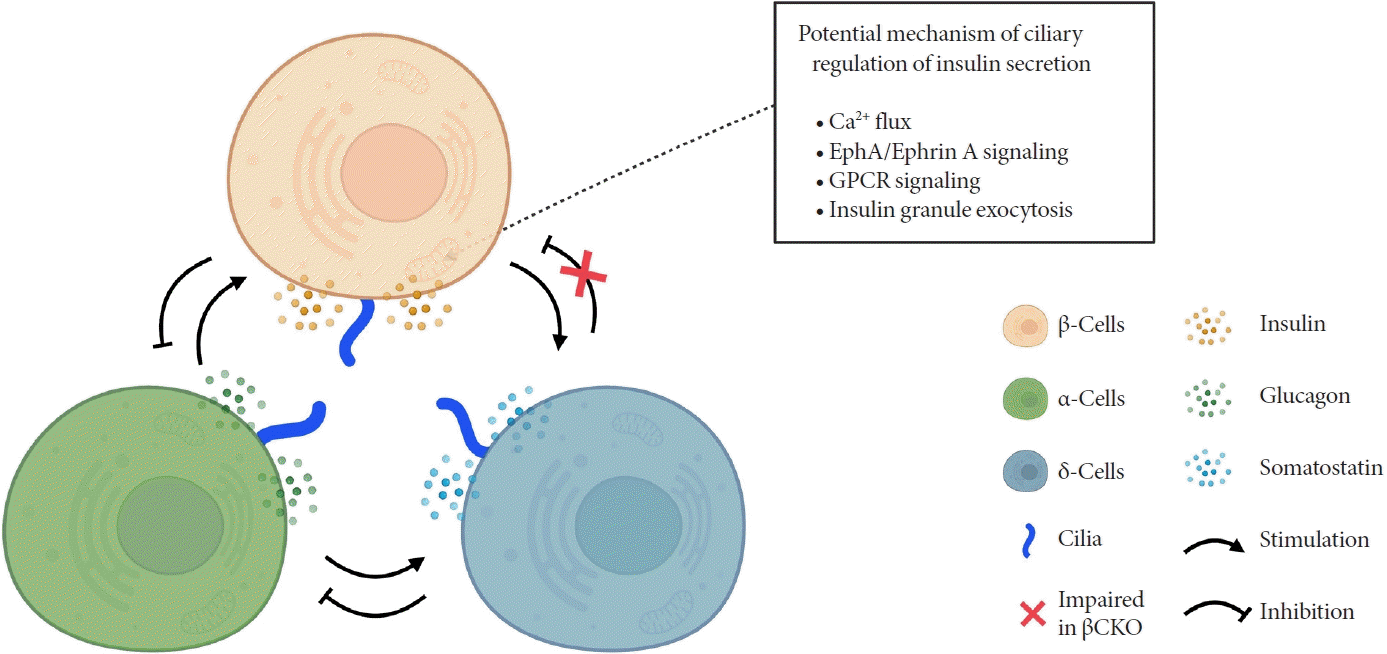
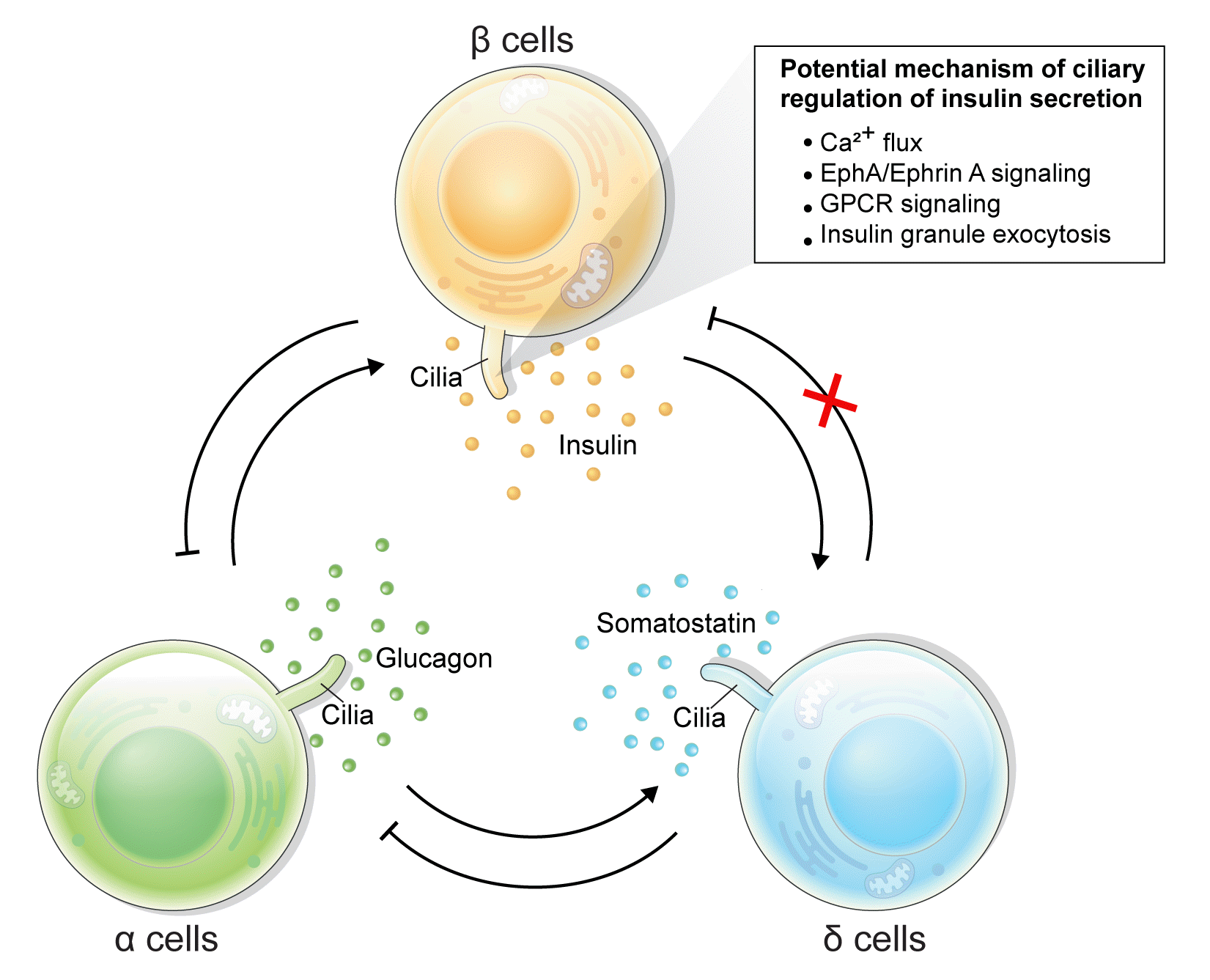
How do cilia regulate insulin secretion? Several key intracellular β-cell events may be under primary cilia control. First, primary cilia regulate glucose-stimulated Ca2+ entry into β-cells. Ift88 deletion and thus the absence of primary cilia in βCKO mouse islets abrogated the first-phase Ca2+ influx as well as second-phase Ca2+ oscillations [42], although it was unaddressed whether these observed effects on Ca2+ are mediated through directly ciliary control of β-cell membrane potential. Second, primary cilia modulate epoxide hydrolase A (EphA)-ephrin-A signaling, a juxtacrine communication pathway that controls insulin secretion in β-cells [85]. Cilia-ablated adult β-cells exhibit hyperphosphorylation of EphA due to impaired endosomal recycling, resulting in Rac family small GTPase 1 (Rac1) inhibition and decrease of insulin secretion [79]. Interestingly, Eph-ephrin signaling may partly account for the reciprocal regulation of cilia and the cytoskeletal network, as actin and microtubule polymerization impact ciliation, and primary cilia signaling through the planar cell polarity pathway may modulate F-actin density, a known mechanism for regulating insulin granule release [86]. There may be direct evidence that cilia loss-of-function causes impaired insulin granule exocytosis, thus accounting for the insulin secretory defect [80]. Bbs4- and oral-facial-digital syndrome type I (Ofd1)-deleted islets had decreased mRNA or protein level of membrane fusion soluble N-ethylmaleimide-sensitive factor attachment receptor (SNARE) proteins Syntaxin1A and synaptosomal associated protein 25 (SNAP-25), while overexpression of Syntaxin1A restored β-cell function comparable to that of the control group [80]. G-protein coupled receptor (GPCR) signaling through the β-cell primary cilium is another established regulator of insulin secretion, a subject that will be discussed in the next section.
Primary cilia of β-cells regulate insulin secretion via cell-intrinsic events as well as via cell communication pathways in response to neighboring islet cells. Direct evidence for primary cilia regulation of paracrine communication has been made in βCKO mouse islets. In the absence of β-cell cilia, δ-cells showed higher basal somatostatin secretion at low glucose and attenuated response to hyperglycemia, revealing the role of β-cell cilia in cross-regulating other endocrine cell types [42]. Inhibition of glucose-induced insulin secretion by somatostatin is impaired in absence of β-cell cilia, while augmentation of glucose-induced insulin secretion by glucagon is intact [42]. These results speak to the highly specialized sensing by the β-cell primary cilium, such that it may selectively mediate responsiveness to one paracrine hormone but not another, both present in high concentrations in the intra-islet space. In support of such specialization, β-cell cilia have been shown to express the somatostatin receptor 3 (SSTR3), as well as the insulin receptor (IR) under dynamic conditions [49,80], but todate there has been no report of ciliary localization of the glucagon receptor. Thus, along with juxtacrine signaling pathways that have been described in β-cell primary cilia, islet cells use their cilia to carry out multiple modes of communication including autocrine, juxtacrine, paracrine, and endocrine, spanning a diverse range of cellular signals. Taken together, β-cell primary cilia play a critical role in crosstalk with α- and δ-cells in maintaining glucose-mediated hormone secretion in the islets (Fig. 3). Given greater degree of heterotypic endocrine cell mingling in human islets than rodent islets [87,88], primary cilia likely play an even more complex role in regulating human islet hormone secretion and maintaining balanced α/β/δ-cell function.
CILIA-DEPENDENT SIGNALING IN PANCREATIC β-CELLS
Primary cilia have vast signaling capacity thanks to its abundant expression of surface receptors, signal transduction proteins, ion channels, and membrane-bound enzymes [6-8]. The enrichment of these molecules within the confines of a small subcellular domain increases the density of signals, making the primary cilium a unique sensor for extracellular cues and intercellular communication [6]. Primary cilia possess a phospholipid composition distinct from that of the plasma membrane, abundant in phosphatidylinositol-4-phosphate (PI(4)P) but deficient in phosphatidylinositol-4,5-bisphosphate (PI(4,5) P2), and localization of phosphatidylinositol-3,4,5-triphosphate (PI(3,4,5)P3) and PI(4,5)P2 at the ciliary base [89,90]. Differential distribution of polyphosphoinositides plays a key role in ciliary protein trafficking and signaling [91]. For example, tubby-like protein 3 (TULP3), a crucial regulator for ciliary trafficking of ion channels and GPCRs, interacts with protein cargo in a PI(4,5)P2-dependent manner [91,92]. The facilitated TULP3-cargo interaction under PI(4,5)P2 in the plasma membrane is weakened in the ciliary membrane lacking PI(4,5)P2. This enables the transport and release of cargo proteins from the plasma membrane to the cilia. Enrichment of PI(3,4,5)P3 and PI(4,5)P2 at the ciliary base contributes to the integrity and barrier function of the transition zone, which controls the entry and exit of ciliary protein [93-95].
A growing number of signaling pathways are coordinated by primary cilia, including sonic hedgehog (Shh), Notch, wingless/Int (Wnt), GPCRs, receptor tyrosine kinases, transforming growth factor-β, and insulin growth factor [6-8]. These signaling pathways have been studied in model organisms such as Drosophila, zebrafish, Chlamydomonas, or Caenorhabditis elegans, while mammalian data comes mostly from in cancer cell lines and mouse embryonic cells [7,8]. Specific to pancreatic cells, one established cilia-dependent signaling pathway is Shh signaling, whose activation is inhibited by primary cilium [82]. Full Shh activation is achieved only under cilium ablation in transgenic mice with pancreas-specific GLI family zinc finger 2 (GLI2) overexpression, a transcriptional mediator of Shh pathway [82]. Insulin signaling pathway via IR is also cilia- and context-dependent. One study has demonstrated that ciliary localization of IR is required for insulin signaling in β-cells, implicating a possible role for cilia in insulin-mediated autocrine or paracrine signaling [80]. In cultured mouse and human β-cells, IR-A is recruited to cilia under insulin stimulation, while basal body/ciliary disruption by Ofd1 or Bbs4 deletion mimics the phenotype of the loss of IR activity in stimulated β-cells [80].
GPCRs represent the largest and most diverse family of signaling receptors in the human body, responding to a variety of external signals including proteins, amino acids, peptides, ions, lipids, nucleotides, photons, and odorants [96,97]. GPCRs are among the most targetable structures for pharmacological modulation as they are accessible on the cell surface and play important roles in human pathophysiology [98]. Many GPCRs are selectively targeted to cilia on various mammalian cells which may confer a spatiotemporal advantage in their signaling [99], as it has been demonstrated that cyclic adenosine monophosphate (cAMP) generation selectively in the cilium in zebrafish and mammalian cells inhibits Shh signaling [100]. Similarly, γ-aminobutyric acid (GABA) receptor B1, recently found on primary cilia of islet cells, exhibits subcellular and even sub-organelle regulation of cilia-dependent signaling [101]. Cilia regulate local Ca2+ influx through GABAB1 receptors while isolating themselves against changes in the cytosolic Ca2+ concentrations through Ca2+ extrusion, and interestingly GABA regulation of ciliary Ca2+ is accomplished through cyclic guanosine monophosphate (cGMP) rather than cAMP [101]. In terms of hormone regulation, two GPCRs, free fatty acid receptor 4 (FFAR4) and prostaglandin E receptor 4 (PTGER4), have highlighted the significance of the cilia-dependent signaling in pancreatic β-cells [9]. Ciliary localization by these GPCRs in β- and α-cells enables ciliary cAMP signaling, and their cognate agonists regulate insulin and glucagon secretion with potencies on par with glucagon-like peptide 1 receptor agonists (GLP-1RAs), one of the most effective insulin secretagogues in clinical use today [9]. There is an additive effect on glucose-induced insulin secretion when FFAR4 agonists are treated with GLP-1RAs [9], suggesting that ciliary GPCR modulation may be an orthogonal therapeutic target to complement existing treatment strategies in diabetes. The SSTR3 is also localized to cilia in pancreatic β-cells [102-104], though the relative contribution of this particular SSTR isoform to the total β-cell paracrine response to somatostatin awaits clarification. Taken together, future investigations in ciliary GPCR signaling in pancreatic β-cells are expected to provide insights into novel therapeutic targets for diabetes and other pancreatic diseases.
CILIARY DYSFUNCTION AND DIABETES
Inherited defects in ciliary genes manifest as genetic ciliopathies in humans. Of these, two representative entities, BBS and Alström syndrome, exhibit prominent metabolic phenotypes including early-onset obesity, insulin resistance, and type 2 diabetes mellitus [105,106]. However, unlike highly penetrant obesity phenotype in both syndromes, the incidence of type 2 diabetes mellitus is significantly higher in Alström syndrome, at 50% to 75% for all ages and up to 82% in those over 16 years of age, compared to less than 25% for BBS [105,106]. In addition, the development of type 2 diabetes mellitus in Alström syndrome appears independent of the onset or severity of obesity [106]. These disparities imply that distinct pathogenesis other than obesity, such as β-cell function or mass, may contribute to diabetes development in affected individuals.
A growing body of evidence from animal and human studies has implicated multiple ciliary proteins in the regulation of metabolism and energy homeostasis. Mouse models of ciliopathy have shown impaired insulin secretion, aberrant islet hormonal regulation, and loss of β-cells over time, with varying degrees of severity and onset timing [40,42,79,80]. Male Bbs4 knockout mice with basal body/ciliary defects exhibit glucose intolerance prior to the onset of obesity [80]. Meanwhile, Ift88 knockout βCKO showed no obesity phenotype but had strong deficits in β-cell insulin secretion and glucose imbalance [42]. Dynamic association of islet cilia with diabetes has been found in diabetes-prone New Zealand Obese (NZO) and diabetesresistant B6.V-Lepobob (B6-ob/ob) mice [47]. NZO islets present fewer cilia under low glucose conditions and exhibit no changes after carbohydrate feeding while well-ciliated B6-ob/ob islets disassemble cilia under high glucose conditions [47]. Islet cilia genes are differentially expressed in B6-ob/ob mice compared to NZO mice and are implicated in cell cycle regulation [47], suggesting that functional cilia dynamics in islets are linked to β-cell proliferation under high glucose conditions and implicating a role for cilia in β-cell mass compensation in type 2 diabetes mellitus.
A number of islet cilia genes become differentially expressed in humans with type 2 diabetes mellitus, consistent with those described in NZO mice [47]. Abnormal glucose metabolism and diabetes phenotypes are described in several human ciliopathy disorders other than BBS and Alström syndrome (Table 3) [105-121]. Individuals with autosomal dominant polycystic kidney disease present abnormal glucose tolerance and decreased insulin secretion [107]. Pericentrin is a pericentriolar molecule that forms a complex with intraflagellar transport and polycystin-2 and is required for primary cilia assembly [122]. A genetic defect in pericentrin (PCNT), a gene encoding the pericentrin, causes early-onset of type 2 diabetes mellitus at an average age of 15 years [108,109]. Increasing depth of transcriptome studies are expected to reveal new ciliary gene linkages to human diabetes [47,48], where the molecular mechanism of individual cilia gene polymorphisms await mechanistic studies and showing the link between genetic variants of ciliary genes and risk of diabetes in humans [123-126].
CONCLUSIONS
The view of primary cilia, once dismissed as rudimentary or “vestigial,” has been changing into one that is all-important for cellular communication and signal transduction. Primary cilia are now regarded as versatile sensors and interpreters of extracellular information, crucial for maintaining homeostasis and cell crosstalk. The accelerated pace of cilia research over the past two decades have led to important discoveries, also raised key questions about cilia action in pancreatic islets. Of particular relevance to islet β-cells, future studies should define the mechanism of nutrient sensing by cilia, how they convey intraislet signals to effect context-specific hormonal secretion, and how cell-to-cell communication might be mediated through cilia, thereby enhancing islet coordination as a unit. Our speculative opinion is that cilia may contribute to β-cell heterogeneity at multiple levels, not only morphologically but also functionally; and are likely controlled via ciliary length, number, motility, and trafficking of ciliary signaling proteins. An intriguing role for cilia in defining functional β-cell subsets was demonstrated in a recent study showing leader β-cells exhibit differential cilia gene expression [127]. Elucidating primary cilia roles in β-cell differentiation, especially in niche stem cell populations, might also inform treatment strategies targeting β-cell regeneration. Given the multi-faceted and dynamic roles that primary cilia play in cellular function and fate determination, future research at the junction of cilia and islet biology should generate new insights to treat human metabolic diseases including diabetes.
 XML Download
XML Download