

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
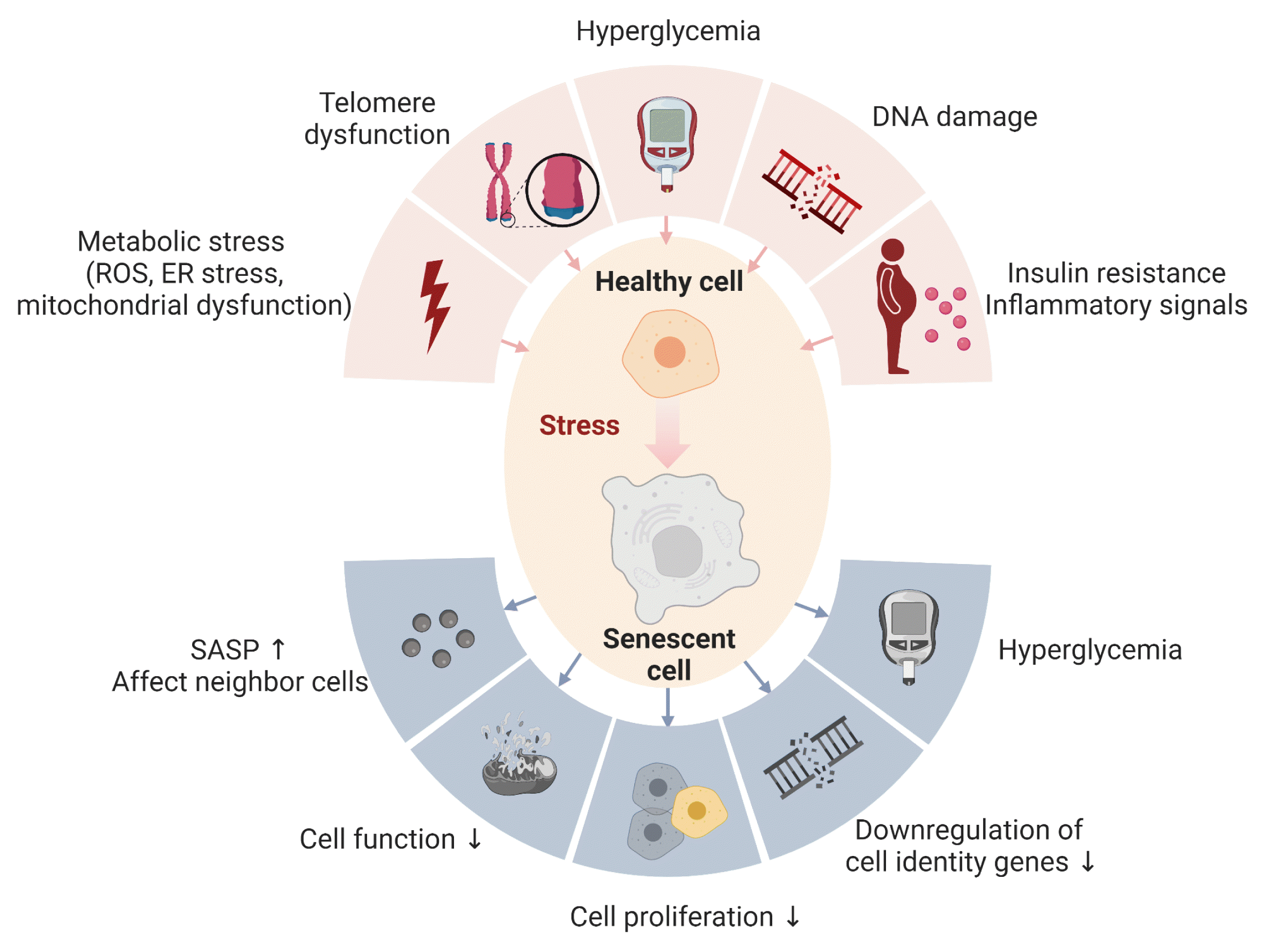
Over the past decade, clinical trials have reported the efficacy of drugs that target cellular senescence and their potential use in the treatment of age-related chronic diseases, such as type 2 diabetes mellitus (T2DM) [1]. When normal cells are subjected to severe DNA damage, they either die by apoptosis or undergo irreversible cell proliferation arrest by induction of cellular senescence. These biological defense mechanisms prevent the proliferation of abnormal cells that have suffered DNA damage. Cellular senescence is the state in which cells irreversibly stop proliferating while retaining their metabolic activity and can be induced by external stressors such as aging, obesity, and radiation due to DNA damage, telomere shortening, and mitochondrial dysfunction. A unique characteristic of senescent cells is the secretion of senescence-associated secretory phenotype (SASP), which induces chronic inflammation through the secretion of inflammatory proteins. The SASP has been involved in the pathogenesis of several age-related diseases, including cancer [2].
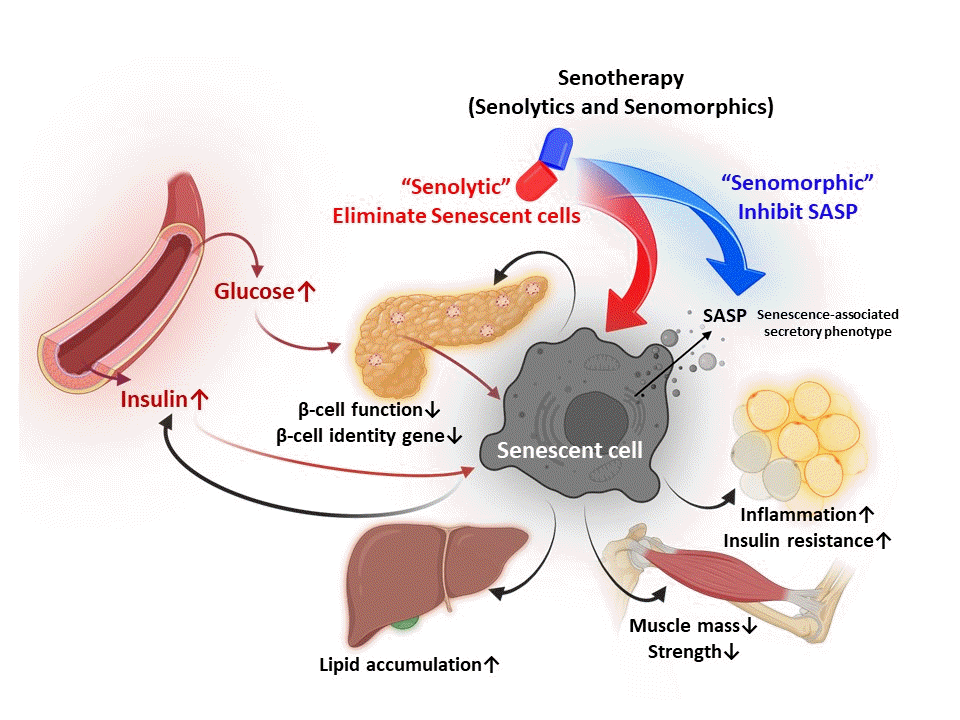
Chronic insulin exposure, which occurs in T2DM, has been shown to cause senescence in hepatocytes [3,4], pancreatic β-cells [5], and adipose tissue (Fig. 1) [6].
Hyperglycemia, pathognomonic of T2DM, can also contribute to senescence through several pathways [7–11], and animal studies have shown that removal of senescent cells improves blood glucose levels and decreases diabetic complications. However, at least two barriers need to be overcome before these therapies can be translated to the clinic: (1) differences between senescent cells in different tissues are unknown, and (2) the specific effects of removing senescent cells in each organ remain to be determined. Therefore, this review focuses on the mechanisms of cellular senescence and its SASP in four key organs for the regulation of blood glucose levels: pancreas, liver, skeletal muscle, and adipocytes, and summarizes ongoing efforts to therapeutically target cellular senescence in them.
The number of people with diabetes in 2021 was estimated to be 537 million and is expected to reach 643 million by 2030 and 783 million by 2045 [12]. Aging is a main risk factor for the development of T2DM; however, currently there are no treatments which target the disease from a cellular aging point of view and existing therapies can manage the condition, but are unable to cure it. Since the discovery of insulin therapy in 1921, various oral and injectable hypoglycemic agents have been developed, and in recent decades, the development of new therapeutic agents such as glucagon-like peptide-1 receptor agonist, dipeptidyl peptidase 4 inhibitors, and sodium-glucose cotransporter-2 inhibitors has expanded treatment options for T2DM. In addition, the complete sequencing of the human genome in 2003 led to the recommendation of personalized medicine for T2DM, which requires an array of information from each person: genetic, physical, and psychological; however, these parameters fail to take into account information at the cellular and molecular level. Therefore, we propose that considering cellular senescence, one of the hallmarks of aging, in the pathophysiology of T2DM could further contribute to a personalized approach to the management of the disease and be a novel therapeutic target.
METHODS
This review is a narrative review; we included the best match articles in PubMed within the last 5 years for each keyword: “senescence,” “pancreas,” “liver,” “muscle,” and “adipose tissue.” and the articles in the references of the subject literature were also included.
CELLULAR SENESCENCE AND SENESCENCE-ASSOCIATED SECRETORY PHENOTYPE
Cellular senescence is an irreversible state of arrested proliferation described by Hayflick and Moorhead [13] in 1961 when they argued that human somatic cells have a predetermined number of divisions (Fig. 1). Hayflick [14] hypothesized that the finite lifespan of diploid cell lines in vitro represented senescence or aging at the cellular level and found that each clonable cell in a population had the same doubling capacity. This was followed by Macfarlane’s 1974 book, in which he coined the term “helical limit” to explain that “normal cells have a finite capacity for replication and cancer cells are usually immortal” [15,16]. It was demonstrated that normal human fibroblasts shorten telomeres as they divide in culture [17], and later in 1988, telomere shortening was shown to cause cellular senescence due to exposed DNA fragments [18,19]. DNA damage activates the DNA damage response system, inducing wild-type activating fragment-1 (WAF1)/cyclin-dependent kinase inhibitory protein-1 (CIP1) and P16 inhibitors of CDK4 (p16Ink4a) cyclin-dependent kinase inhibitors, leading to cell cycle arrest. In addition to growth arrest, senescent cells also undergo morphological flattening in culture and the generation of reactive oxygen species favor the activity of the senescence-associated β-galactosidase (SA-β-gal) [20,21]. β-Gal is a hydrolytic enzyme localized in lysosomes. Normal cells express endogenous β-gal whose optimal activity occurs at pH 4.0; in senescent cells, intralysosomal pH changes to 6.0 increasing the activity of SA-β-gal which is used as a marker for senescent cells. However, it is not a specific marker and must be used in combination with others [22,23].
Age decreases autophagy which is a process by which a cell breaks down and destroys old, damaged or abnormal proteins in the cytoplasm and requires lysosomal integrity [24]. It is reported that the transcription factor MondoA may delay cellular senescence by activating autophagy through inhibiting Rubicon, an autophagy-negative regulator [25,26]. Further studies to understand the pathways linking lysosomal changes in senescence and autophagy are needed.
As mentioned, there are no universal markers of senescence, therefore it is recommended that multiple hallmarks of senescence are used to evaluate potentially senescent cells; these markers can be divided by the different traits displayed by this cell population: cell cycle arrest (p16Ink4a, p21Cip1, p53, Rb, pRb), SASP factors (serpine 1, interleukin 6 [Il6], Il1a, tumor necrosis factor α [Tnfα], CC motif chemokine ligand 3–5 [Ccl3–5], etc.), nuclear reorganization or DNA damage (P53BP1, γH2AX, lamin B1, telomere associated foci), changes in lysosomal compartment (SA-β-gal activity, lipofucsin) [19].
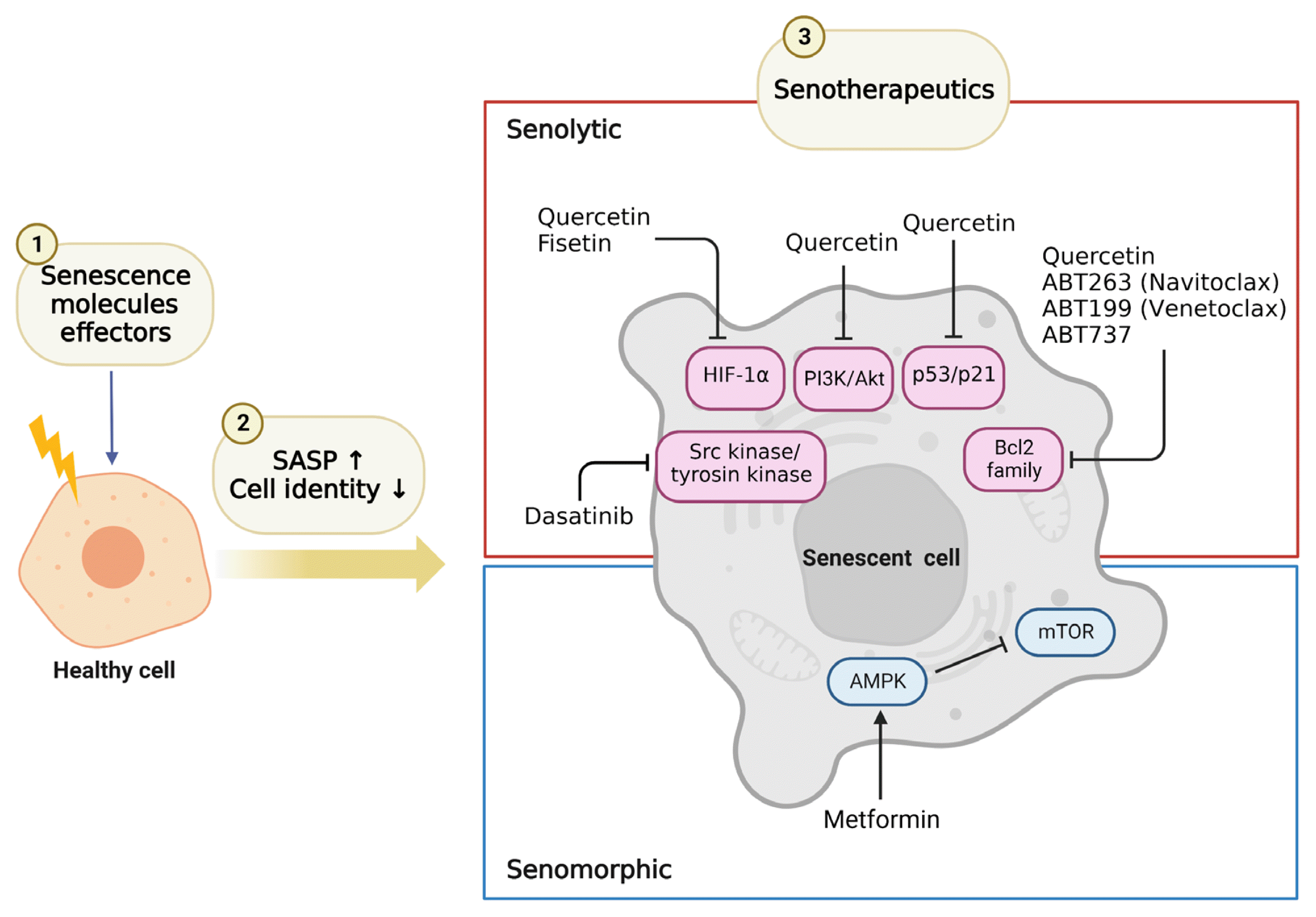
Another key characteristic of senescent cells is their resistance to apoptosis [27] due to the upregulation of senescent cellular anti-apoptotic pathways (SCAP), such as B-cell lymphoma 2 (BCL-2)/BCL-XL, phosphoinositide 3-kinase (PI3K)/AKT, p53/p21/serpines, dependence receptors/tyrosine kinases, and hypoxia-inducible factor 1α (HIF-1α). Drugs or interventions that target these pathways have led to the development of senolytic drugs which induce apoptosis of senescent cells. This topic will be discussed in more detail in the ‘Senotherapy’ section.
Some senescent cells secrete the SASP consisting of various factors such as inflammatory interleukins, chemokines and growth factors and extracellular matrix-degrading enzymes, which were originally identified using antibody arrays and unbiassed proteomic of conditioned media obtained from senescent cells (Fig. 1) [28]. Representative molecular mechanisms that regulate SASPs include the P16INK4A and P21WAF1/CIP1 pathways, DNA damage response signaling, the nuclear factor-κB and the p38/mitogen-activated protein kinase (MAPK) pathway, mammalian TORC1 (mTOR), the Janus kinase/signal transducer and activator of transcription 3 (JAK/STAT3) IL-1α, and cyclic GMP-AMP synthase (cGAS)-stimulator of interferon gene (STING) pathways [29–34].
The physiological role of local SASP secretion is thought to be recruitment of immune cells to eliminate senescent cells which cannot undergo apoptosis due to upregulation of SCAPs. However, deleterious effects of SASP have also been described such as promoting local inflammation, inducing local dysfunction and early entry into senescence of healthy cells and support of cancer cell growth in the microenvironment [35].
Secretion of SASP factors is a crucial step during wound healing. During the repair of damaged skin tissue, SASP is transiently produced by fibroblasts, which attracts immune cell for clearance of damaged tissue cells. At the same time, senescent fibroblasts generate growth factors that induce the proliferation and differentiation of skin progenitor cells, resulting in the production of new skin. By the time tissue repair is complete, senescent cells causing SASP are cleared [36]. However, sustained long-term secretion of SASP results in a cancer-promoting environment [37,38]. Additionally, SASP factors have also been shown to induce cellular senescence in surrounding cells in a paracrine manner and to enhance their own cellular senescence phenotype in an autocrine manner [39].
While senescent cells function as a critical cancer suppression mechanism in mammals, they can have deleterious effects as they accumulate in various tissues with age and may contribute to the development of age-related diseases [40] since immune function declines with age, resulting in an increased load of senescent cells [41]. Animal studies have been conducted to understand whether the elimination of senescent cells could lead to new treatments for age-related diseases. Several genetically engineered mice have been created where senescent cells can be selectively removed and are optimal for studying the relationship between cellular senescence and age-related diseases. Various clinical trials using senolytics are also currently under way (Table 1).
In the next section, we will summarize the potential contribution of cellular senescence to T2DM and the studies of cellular senescence-depleting agents in various organs for potential future therapeutic strategies.
HYPERGLYCEMIA AND SENESCENT CELLS
Hyperglycemia increases cellular senescence through several mechanisms (Fig. 1). Telomere shortening is enhanced in diabetic patients, suggesting a potential link between cellular senescence signals and the development of diabetes [42].
Additionally, hyperglycemia induces senescence in human dermal fibroblasts through cell migration [43] and in endothelial cells through the aquarius intron-binding spliceosomal factor/plasminogen activator, urokinase (AQR/PLAU) signaling axis [7]. Since senescence pathways induced by hyperglycemia are tissue-specific, they will be discussed separately in the setting of T2DM.
CELLULAR SENESCENCE IN PANCREATIC β-CELLS
Insulin resistance induces β-cell senescence, which can contribute to the development of T2DM (Fig. 1) [5]. One of the first reports of increased β-cell senescence in a high-fat diet (HFD) model was done by Sone and Kagawa [44] suggesting that it might be a vital contributor to the pathogenesis of T2DM. By using the p16Ink4a apoptosis through targeted activation of caspase (INK-ATTAC) mouse model [45], where p16Ink4a+ cells can be selectively cleared from the whole body, we showed that targeting this senescent cell population improved glucose metabolism and insulin secretion, decreased expression of aging, senescence, and SASP genes in islets from aging and insulin resistance models. Additionally, administration of ABT263 (Navitoclax), a BCL-2 inhibitor used as an oral senolytic, improved hyperglycemia and β-cell gene expression profiles in animals treated with the insulin receptor antagonist S961. However, there are no reports of the clinical application of ABT263 as a senolytic drug, and further studies are required to reduce its toxicity. Other known senolytics include dasatinib (D) (tyrosine kinase inhibitor which targets several SCAPs: p53, p21, ephrin receptors) and quercetin (Q) (flavonoid which targets the PI3K, AKT, mouse double minute 2 homolog [MDM2], P53 and HIF-1α SCAP pathways). In combination, dasatinib and quercetin (D+Q) reduce senescent cells [46–50]. Mice with S961-induced acute insulin resistance received Q (50 mg/kg) and D (5 mg/kg) once a week, and showed improvement in blood glucose levels. Human β-cells also showed the same biological features of increased cellular senescence and overexpression of P16INK4A with aging and T2DM [5]. The molecular basis of pancreatic β-cell senescence remains to be determined and could lead to the identification of new therapeutic targets.
CELLULAR SENESCENCE IN THE LIVER
It has been reported that nonalcoholic steatohepatitis (NASH)/nonalcoholic fatty liver disease (NAFLD) status increases the risk of developing T2DM [51], and people with diabetes are reported to be at increased risk of developing both NAFLD to NASH [52].
Fat accumulation in hepatocytes promotes telomere shortening and DNA damage, which may induce hepatocyte senescence [21,53]. Aravinthan et al. [53] showed that compared to normal human hepatocytes, hepatocytes from humans with NAFLD have shorter telomere length and cell-cycle arrest after the G1/S phase and high expression of p21Cip1. Hepatocyte senescence is associated with liver fibrosis and diabetes. Prolonged culture of human hepatocytes under hyperglycemia increased p53 and p21Cip1 expression while inhibition of the insulin signaling pathway prevented cellular senescence [54]. The PI3K/AKT/mTOR signaling pathway is required to induce senescence markers which can be blocked by inhibiting insulin signaling. D+Q reduced overall hepatic steatosis in aged, obese, and diabetic mice [55]. Therefore, the combination treatment of D+Q antagonizes hyperinsulinemia-induced senescence in hepatocytes and reduces the expression of SASP factors, implying that senolytic therapy may be beneficial for alleviating hyperinsulinemia-related liver senescence and associated complications [54]. Impairment of mitochondrial function and lipid metabolism in senescent hepatocytes is a major driver of hepatic steatosis and could explain its progression of NAFLD [55]. Liver biopsies of 58 patients showed that hepatocyte senescence was strongly associated with the severity of NAFLD/NASH. Bone morphogenetic protein 4 (BMP4) exhibited anti-aging, anti-steroidal, anti-inflammatory, and anti-fibrotic effects, while Gremlin 1, particularly abundant in human visceral fat, was found to be anti-aging and antagonistic to BMP4. Both target the yes-associated protein (YAP) and the transcriptional coactivator with PDZ-binding motif (TAZ) pathway, a likely a regulator of senescence and its effects [56].
CELLULAR SENESCENCE IN MUSCLE
Cellular senescence has been reported within all muscle types: cardiac [57,58], smooth [59,60], and skeletal [61,62]. Common markers of senescence are prevalent in these tissues, like elevation of p21Cip1 and p16Ink4a, increased SA-β-gal activity, and the presence of SASP factors. The high concentration of mitochondria within muscle cells can expose them to greater amounts of reactive oxygen species (ROS), a known inducer of senescence, which can lead to conditions that push naïve cells into a senescent state. Treatment with D+Q reduced senescent markers in mice with hypercholesterolemia and improved vasomotor function in cardiac muscle, while treatment with ABT263 cleared senescent cells after myocardial infarction [57,63]. Vascular smooth muscle cells (VSMCs) are particularly susceptible to ROS, SASP, and DNA damage, which can lead to chronic inflammation and accumulation of senescent cells, a possible link to the development of atherosclerosis. Treatment with metformin, an anti-diabetic medication with anti-senescence properties, improved vascular function by enhancing mitochondrial function, and treatment of cultured VSMCs with Q reduced activation of p21Cip1 and p16Ink4a pathways [64,65].
Within skeletal muscle, extensive use and high-force contractions from resistance training produce mechanical and chemical stress, changes in Abbreviation is adenosine monophosphate (AMP)/adenosine triphosphate (ATP) ratios, changes in calcium concentrations, and decrease in the partial pressure of oxygen, all activating pathways associated with senescence [61,62]. Skeletal muscle cells, skeletal muscle stem cells or satellite cells, and mesenchymal muscle progenitors, known as fibro/adipogenic progenitors, can undergo senescence [61,62,66].
Several studies have targeted senescent cells in skeletal muscle. ABT263 was shown to reduce SASP in a mouse model of Duchenne muscular dystrophy (Mdx mice), while an NAD+ precursor nicotinamide riboside decreased SASP in muscle stem cells and prevented senescence in Mdx mice and aged mice [67]. In addition, D+Q improved treadmill endurance in a mouse model of one-leg irradiation and improved bone parameters in an excision repair cross-complementation group 1 (Ercc1)−/Δ mouse model of accelerated aging [48].
Age-related loss of muscle mass and strength is termed sarcopenia [68]. In satellite cells from old people with sarcopenia, P16INK4A was reported to be positively correlated with decreased myogenesis and increased senescence cells [69]. In skeletal muscle tissue, inflammation-induced cytokines such as TNFα, IL-18, and IL-6 are believed to induce muscle atrophy during sarcopenia [70,71] and are hallmark SASP factors.
CELLULAR SENESCENCE IN ADIPOCYTES
Cellular senescence has been reported in mouse adipose tissue [72–74], and human preadipocytes [33,49,75,76]. The cell cycle progresses when adipocytes are exposed to continuous hyperinsulinemia in vivo or in vitro. Furthermore, a senescent cellular program is initiated in mature adipocytes. Adipocyte senescence is associated with SA-β-gal staining, loss of nuclear high mobility group box 1 (HMGB1) protein, high expression of cyclin D1, increased cell and nuclear size, p21Cip1 and p16Ink4a expression, and a non-resolving continuous DNA damage response visualized by γH2AX staining, all features of early senescence [6].
Mature adipocytes were thought incapable of re-entering the cell cycle but it has been reported that continuous exposure to hyperinsulinemia in vivo or in vitro causes cell cycle progression and a concomitant increase in DNA content, activation of the senescent cell program, and inflammatory SASPs leading to human adipose tissue inflammation [6]. While it has been reported that senolytics improved patients’ physical function [77], the first clinical trial demonstrating that senolytics reduce human senescent cells at the cellular level was reported in 2019. Patients with diabetic nephropathy were treated with D+Q for 3 days, and senescence of abdominal subcutaneous adipose tissue and skin epidermis were compared before and 11 days after the treatment. In both abdominal subcutaneous adipose and skin tissues, P16INK4+ and P21CIP1+ cells were decreased, and circulating SASP factors such as IL-1α, IL-6, matrix metallopeptidase 9 (MMP-9), and -12 were decreased in the epidermis [49]. SA-β-gal activity in subcutaneous adipose tissue was positively correlated with serum leptin, markers of insulin resistance, and increased trunk fat mass, but not body mass index or age [78].
SENOTHERAPY: SENOLYTICS AND SENOMORPHICS
Senotherapies may reduce the onset of age-related pathologies and they are mainly composed by senolytics and senomorphics. Senolytics selectively kill the senescent cells by targeting SCAP pathways, and RNA interference approaches have identified drugs that target nodes in key SCAP, such as BCL-2/BCL-XL, PI3K/AKT, p53/p21/serpions [48,27]. By using this approach, several senolytics (induce apoptosis of senescent cells) have been discovered. Senomorphics target SASP pathways without killing senescent cells. This section will focus on senolytics and senomorphics used in clinical trials and animal research, which are relevant to T2DM and its complications (Fig. 2). D, Q, fisetin, and ABT263 have been used as senolytics, while metformin has been used as a senomorphic. Clinical studies targeting senescent cells in diabetes mellitus, diabetic complications, or relevant organs are summarized in Table 1.
DASATINIB AND QUERCETIN
D is a tyrosine kinase inhibitor [46] and is used in chronic myeloid leukemia and Philadelphia chromosome-positive acute lymphoblastic leukemia that is resistant or intolerant to therapy. Q is a natural flavonoid and inhibits PI3K [47]. The first senolytic used clinically was the combination of D+Q, reported in 2015 [48]. In insulin resistance using S961, an insulin receptor antagonist, we reported that D+Q reduced the number of β-gal-positive dispersed islet cells in vitro and in vivo, and it reduced blood glucose in a model of insulin resistance in mice [5].
The combination of D+Q decreased the number of naturally occurring senescent cells and their secretion of SASP in human adipose tissue explants. Greater omental adipose tissue obtained by gastric bypass surgery from eight obese subjects was used [79]. Side effects of D+Q include hematologic dysfunction, fluid retention, skin rash, and QT prolongation [80]. Further clinical studies in a population of T2DM are needed to evaluate its potential efficacy in this disease.
BCL-2 INHIBITORS: ABT263 (NAVITOCLAX), ABT199 (VENETOCLAX), AND ABT737
ABT263 (Navitoclax) targets the BCL-2 pathway [81]. We reported that ABT263 killed a significant portion of a β-gal-positive subpopulation of dispersed islet cells in vitro. In addition, ABT263 reduced blood glucose levels and p16Ink4a expression levels in a drug-induced insulin-resistant mouse model. Also, peripheral tissues had significantly decreased p16Ink4a in a model of HFD-induced insulin resistance [5]. In combination with D+Q, Navitoclax selectively eliminated senescent cells and reduced lung disease in severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2)-infected hamsters and mice [82]. However, diarrhea, nausea, and thrombocytopenia are common side effects, limiting its clinical use.
ABT199 (Venetoclax) selectively inhibits BCL-2, which is an apoptosis suppressor protein [83]. Venetoclax is an oral BCL-2 inhibitor approved for relapsed/refractory chronic lymphocytic leukemia and for acute myeloid leukemia. Administration of ABT199 to normoglycemic non-obese diabetic (NOD) mice suppressed diabetes development compared to controls in a model of type 1 diabetes mellitus (T1DM) [84].
SENOMORPHIC: METFORMIN
Metformin has been used for decades to treat T2DM and lower circulating glucose levels by inhibiting hepatic gluconeogenesis [86]. Additionally, metformin may have anti-aging properties by preventing DNA damage and inflammation [68] and has been shown to have senomorphic effects [87]. The effects of metformin have been reported in a mouse model of kidney disease where a brief pulse inhibited acute senescence of bone marrow mesenchymal stem cells (MSCs) [88]. Others have shown that metformin reduces ROS levels and initiation of senescence in mouse adipose-derived MSCs [89]. Treatment of human adipose stem cells with metformin in therapeutic concentrations for 6 weeks resulted in reduced levels of β-gal activity [90]. Measuring circulating SASP before and after the administration of metformin would potentially identify patients who would benefit most from metformin’s senomorphic effects.
OTHERS: CURCUMIN
Curcumin is a phytocompound in the root of turmeric used in many clinical trials in T2DM. The combination of the antioxidant polyphenolic compounds curcumin and hesperetin has been reported to improve cellular senescence in D-galactose-induced senescent neurons and rats, including reduced β-gal staining cell number, p16Ink4a, and p21Cip1 [91]. Curcumin has also been reported to improve D-galactose-induced cellular senescence in cardiomyocytes by promoting autophagy through the sirtuin 1 (SIRT1)/AMP-activated protein kinase (AMPK)/mTOR pathway [92].
Although many clinical studies have been reported on curcumin in T2DM, there is still no report that has seen a direct effect on senescent pancreatic β-cells. Many clinical trials using curcumin showed a significantly reduced T2DM incidence among people with prediabetes. In addition, curcumin treatment in a diabetic mouse model improved β-cell function with higher homeostasis model assessment of β-cell function (HOMA-β) and lower homeostasis model assessment of insulin resistance (HOMA-IR) compared to the placebo group [93]. It has been reported that diabetes-induced pathological changes in the aorta are protected by curcumin, mainly through inhibition of JNK2, accompanied by upregulation of nuclear factor erythroid 2-related factor 2 (Nrf2) expression and function [94]. Additionally, more myocardial protection was evident with the combination of metformin and curcumin than with metformin alone in diabetic rats, suggesting that inhibition of the JAK/STAT pathway and activation of the Nrf2/heme oxygenase 1 (HO-1) pathway may be among the mechanisms mediating the effects of curcumin and metformin [95]. Interestingly, the JAK/STAT pathway is also a known regulator of SASP secretion, making this a potential therapeutic mechanism. Although there are no reports yet of clinical studies in diabetic patients using curcumin as a senescence cellular remover, further accumulation of research using this compound is warranted.
CONCLUSION AND FUTURE DIRECTIONS
Hyperglycemia and hyperinsulinemia, both hallmarks of T2DM, lead to senescence in pancreatic β-cells and other relevant tissues like liver, thereby contributing to disease progression and its complications. Therefore, senotherapeutics might be a novel treatment for this age-related disease.
The use of senolytics to differentially target senescent cells in each organ is currently unavailable and is an active field of research. If achieved, it could contribute to personalized care for people with different subtypes of T2DM as described by Ahlqvist et al. [96].
Besides using drugs to target senescent cells, it is important to consider how lifestyle interventions, such as diet and exercise, affect senescence. For example, exercise is an effective strategy to prevent cellular senescence and extend life expectancy and health span; since T2DM is a disease of accelerated senescence cells, exercise may be an effective treatment with anti-aging effects [97]. The Mediterranean diet has been associated with improved health in old age and a decrease on the nine hallmarks of aging [98] and its effects on the management of senescence in T2DM should be further researched.
Recent advances in other fields should also increase our understanding of cellular senescence and its role in disease. For example, the completion of the human genome sequencing including telomere regions [99] will shed light on the particular contributions of these chromosome structures in determining the load of senescent cells in a given tissue.
Currently, cellular senescence is a very active field of research and many expectations revolve arounds its role in age-related diseases. In T2DM, experimental and pathological studies support a role of this targetable cellular process in the disease, but caution must be observed as these findings are validated in the clinical setting and its therapeutic applicability. Continued rigorous studies and consortium efforts [100] will increase our understanding of senescence biology and contribute to developing effective therapeutics leading to optimal and personalized interventions.
 XML Download
XML Download