

 PDF
PDF Citation
Citation Print
Print



서 론
Primitive myxoid mesenchymal tumor of infancy (PMMTI)는 2006년 Alaggio 등[1]이 처음으로 발표한 소아의 연조직(soft tissue)에 발생하는 매우 드문 악성종양이다. 이 악성종양은 신생아부터 발견될 수 있으며 주로 1세가 되기 전에 체간, 사지, 두경부 등 다양한 영역에서 발견된다. 국소적으로 매우 공격적인 질환으로 알려져 있으나 아직 그 사례가 드물어 자세한 임상경과를 알기 힘들다[2]. 현재까지의 치료방법으로는 수술적 치료를 통하여 완전 절제를 하는 것이 원칙이나, 절제하더라도 병변의 재발 및 타 부위로 전이가 관찰될 수 있다[3]. 절제를 할 수 없는 경우에는 항암요법, 방사선요법 등의 치료를 시도할 수 있으나, 이마저도 절반 정도에서는 치료에 실패한다[4]. 저자들은 우연히 발견된 후두부(occipital area)의 종물을 주소로 내원한 3세 여아 PMMTI 사례를 경험하였기에 이에 대한 임상 경과 및 치료 결과에 대해 보고하고자 한다.
증 례
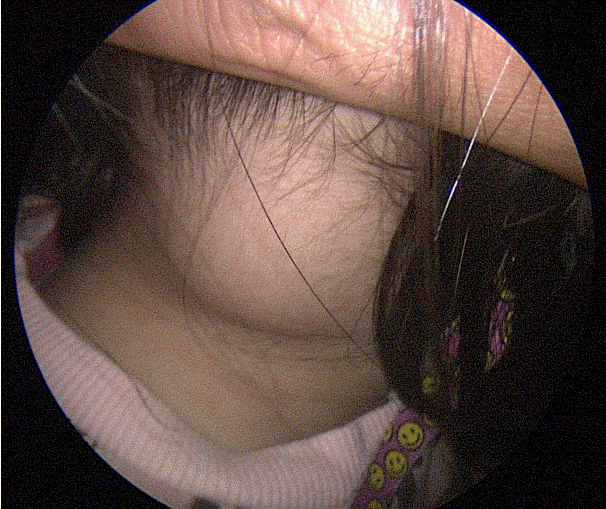
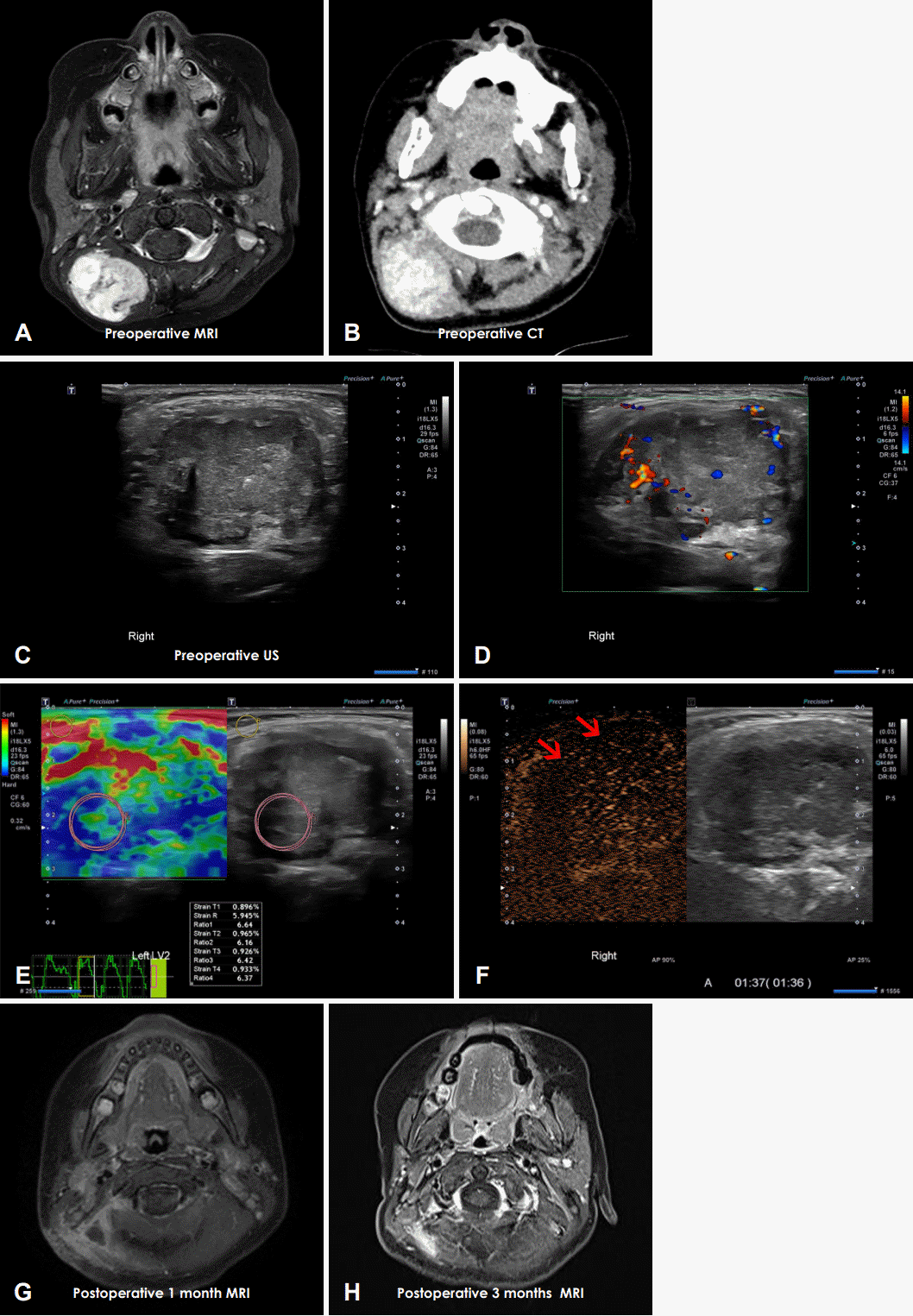
3세 여아가 11개월 전 우연히 만져진 우측 후두부의 종물을 주소로 내원하였다(Fig. 1). 종물이 점점 커지는 양상으로 발견 1개월째에 타 병원에 내원하여 시행한 초음파상 우측 후두부에서 2.8×2.3×1.8 cm의 낭성 변화(cystic change)가 있는 종양성 병변이 관찰되어 세침흡인검사(fine needle aspiration)를 시행하였으나, 비진단 소견(non-diagnostic)으로 경과 관찰 하였다. 발견 6개월째 재내원하여 시행한 초음파상 3.1×3.8 cm의 비균질형 동에코 종양 소견(heterogeneous isoechoic mass)으로 크기가 증가했고 경부 자기공명영상(MRI)상에서는 우측 후두부에 2.7×2.8×3.8 cm의 T1 고신호강도(high signal), T2 비균질 고신호강도(heterogeneous high signal) 소견으로 연조직 육종(soft tissue sarcoma)이 의심되지만 두개저의 뼈 침범은 관찰되지 않았다(Fig. 2A). 발견 10개월째 타 병원에서 절제 생검을 계획하였으나 환자 사정상 시행하지 않고 초음파 유도하 중심부바늘생검(core needle biopsy)를 시행하였으며, 악성도가 확실하지 않은 중간엽 종양(mesenchymal tumor) 소견을 보여 추가적인 평가 및 치료를 위해 본 기관으로 의뢰되었다.
종물 발견 11개월째가 되는 본원 내원 당시 우측 후두부에 약 4 cm 크기의 단단한 무통의 종물이 촉진되었으며, 경부 컴퓨터단층촬영(CT)상 약 3.8×3.0×3.7 cm의 비균질성의 조영 증강이 뚜렷한 종양(heterogeneous and avidly enhancing mass)이 우측 후두부에서 관찰되며 주변부의 뼈 침식이나 주변 지방 침윤(perilesional fat infiltration)은 관찰되지 않았다(Fig. 2B). 초음파상에서는 우측 후두부에 약 4.0×2.7 cm 크기의 단단한 종양이 관찰되며 병변의 주위로 혈관 분포(peripheral vascularity)가 보였다(Fig. 2C and D). 종양의 강직도(stiffness)를 알아보기 위하여 시행한 변형 탄성 초음파(strain elastography)상 종양의 상대적인 변형률(strain ratio)은 평균 6.4로 피하 및 등세모근 근육과 비교하였을 때 약 6.4배 단단한 소견을 보였다(Fig. 2E). 조영증강초음파(contrast enhanced US)에서는 병변의 표층(superficial portion)에서 관류 결손(perfusion defect)이 보여 내부 괴사(internal necrosis) 혹은 점액성 요소(myxoid component)가 있을 것으로 판단되었다(Fig. 2F). 본 기관에서 다시 한번 시행한 초음파 유도하 중심부바늘생검상 PMMTI에 가장 근접하나 영아 섬유 육종(infantile fibrosarcoma)을 배제할 수 없다는 병리 결과를 확인 후 수술적 절제를 계획하였다.
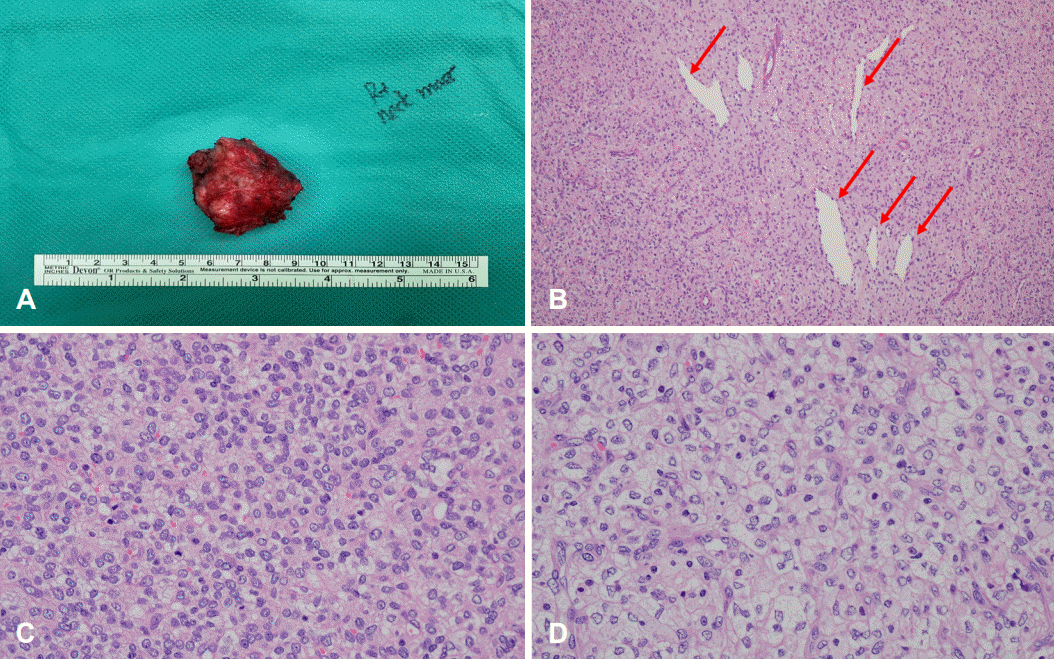
수술실에서 확인 당시 우측 후두부에 약 4 cm 크기의 종물이 등세모근보다 깊게 위치하고 있었고 등세모근과 유착이 있어 일부 근육을 포함하여 종물을 박리하였다. 수술 당시 육안상 종물이 주위 조직과 유착이 있어 보였기 때문에 주위 조직을 포함하여 종물을 파열 없이 제거하였다(Fig. 3A).
술후 조직검사 결과상 PMMTI가 진단되었고 종물의 크기는 3.7×3.2×2.2 cm으로 측정되었다. 병리 검사상 100배 배율에서 종양의 다양한 부분에서 혈관주위세포종 양상의 혈관(hemangiopericytoma-like vessel)이 관찰되었다(Fig. 3B). 400배 배율에서 타원형 또는 원형의 핵 및 수포성 염색질 형태(vesicular chromatic pattern)가 보이며 연한 호산성 종양세포질(pale eosinophilic tumor cytoplasm)을 보인다(Fig. 3C and D). 종물을 파열 없이 제거하였기 때문에 병리 결과상 절제연 음성(margin negative)이 나올 것으로 기대하였으나 절제연 양성(margin positive) 소견이 나왔고, 보호자에게 재수술 필요성 설명하였으나 주변 구조물의 광범위한 절제를 당장 원하지 않고 추적 관찰을 원하여 소아 혈액종양내과와 상의하여 단기간 영상 추적 관찰하면서 필요 시 재수술 및 추가 항암치료를 고려하기로 하였다. 술후 1개월에 시행한 MRI상 수술 부위로 액체 저류 및 조영 증강되는 병변이 관찰되었으나(Fig. 2G), 수술 3개월에 시행한 MRI T1 강조 이미지에서 해당 부위의 전체적인 조영 증강이 감소한 소견을 보이며 명확한 종물 양상의 병변이 보이지 않았다. 수술 6개월 시점에도 신체 진찰상 우측 후두부에 종물 등 특이소견을 보이지 않아 경과를 지켜볼 예정이다(Fig. 2H).
고 찰
PMMTI는 기존까지 미분화 육종(undifferentiated sarcoma)으로 분류되거나 선천성 영아 섬유육종으로 분류되었다. 하지만 두 육종과는 다르게 원시 미성숙 중간엽 세포(primitive mesenchymal cell)가 세포학적 이형성증을 갖는 점액체(distinctive myxoid background) 내에서 자란다는 특징을 가지며 더 공격적인 임상 경과를 나타낸다는 점으로 2006년 Alaggio 등[1]이 발표한 논문에서 PMMTI라는 용어가 새롭게 사용되었다. PMMTI의 진단 기준은 아직 정해지지 않았으나, 기존에 보고된 사례들의 현미경 소견을 종합하면 종양세포는 원시 방추형(primitive spindle), 다각형(polygonal) 또는 원형의 모양을 가질 수 있으며 종양세포의 핵은 단조로우면서 동일한 형태를 보이며 핵의 염색질은 균일하고 핵소체는 거의 관찰되지 않는다. 이러한 종양세포들은 점액성 기질(myxoid stroma)을 기반으로 성장하며 고세포충실도와 저세포충실도를 모두 보이는 경우가 많다. 종양세포 덩어리 사이로 혈관 증식이 많으며 혈관주위세포종 양상의 혈관 모양도 자주 관찰되며 저배율상에서 종양은 결절형으로 자라는 경우가 많으며, 각각의 결절들은 콜라겐 기질로 구분된다.
PMMTI의 치료는 아직 정립되어 있지 않지만 2020년 Asaftei 등[4]이 발표한 논문에 따르면 2006년부터 2020년까지 PMMTI는 총 29예가 발표되었으며 이 중 치료 방법을 알 수 없는 10건을 제외하면 17예에서 수술을, 1예에서 항암치료를, 1예에서 수술과 항암치료를 병행하였다. 이 중 수술 후 결과를 알 수 없는 1건을 제외한 총 17예의 수술 사례 중 65% (11/17)에서는 술후 잔여 병변이 있었으며 35% (6/17)에서는 잔여 병변없이 제거가 이루어졌다. 잔여 병변이 있는 11예 중 8예에서는 재발하였으며(73%) 이 중 2건에서는 2차 치료로 항암치료를 하였으나 재발하여 수술적 치료를 하였고[1,5], 2건에서는 2차 치료로 수술과 항암 병합 요법을[1,6], 4건에서는 수술을 하였다[1,3,7,8]. 잔여 병변 없이 제거가 이루어진 6예에서는 1건만이 재발하였으며(17%) 2차 치료로 재수술을 하였으나 다시 재발하여 치료 없이 경과 관찰 하였다[3]. 항암치료를 단독으로 시행한 경우는 2건의 사례가 있었으며 Cuthbertson 등[9]은 1차 항암치료 후 재발하여 2차 치료로 수술을 하였으나 재발하여 3차 치료로 수술 및 항암치료를 병행하여 재발하지 않았고 Memmott 등[10]은 항암치료 후 병변이 재발하지 않았다고 보고하였다.
PMMTI의 수술 후 평균 재발 기간은 4.4개월(1-12개월)로 매우 빠르며 수술 후 잔여 병변이 남아있는 경우에도 더 이상 크기 변동 없이 안정적인 상태를 유지하는 기간은 평균 25.3개월(5-70개월)이다[4]. 우리 증례의 경우 술후 1개월에 시행한 MRI상 수술 바닥에 액체 저류 및 조영 증강 관찰되었으나 수술없이 경과 관찰 하였고, 수술 3개월에 시행한 MRI상 수술 1개월에 보이던 액체 저류는 없어졌고 전체적인 조영 증강이 감소한 소견을 보였다. 하지만 평균 재발 기간이 4.4개월임을 감안하고 1년 뒤에 재발한 사례도 있는 것을 감안하였을 때, 단기간 및 장기간 경과 관찰은 필수적일 것으로 보인다.
앞서 언급한 수술 후 잔여 병변이 보이지 않는 6예 중 재발이 발생하지 않은 5예의 공통점은 수술 후 병리 검사상 현미경상 잔여 병변의 존재가 없었다는 점이다[1,11-14]. 재발이 발생한 1예에서는 경부에 발생한 3 cm의 병변으로 수술 후 육안적으로는 잔여 병변이 없었으나 현미경상 잔여 병변의 유무를 확인하지 못했던 사례였다[3]. 따라서 본 증례의 경우에도 충분한 수술 절제연의 확보가 되지 않았기 때문에 원칙상으로는 추가적인 절제 및 보조 방사선요법 등의 치료를 하는 것이 예후가 더 좋을 수 있으나, 환자 및 보호자와 상의하여 밀접한 경과 관찰을 시행하기로 하였다.
결론적으로 PMMTI이 절제가 가능한 부위에 있고 첫 번째 치료로 수술적 치료를 선택했을 경우 현미경상 잔여 병변이 없이 절제하는 것이 최상의 선택으로 보이며 수술 시 종물을 피막에 붙여서 절제하는 것보다는 경계를 충분히 추고 절제를 하는 것이 재발을 줄이는 데 도움을 줄 수 있을 것으로 보인다.
 XML Download
XML Download