

 PDF
PDF Citation
Citation Print
Print



Introduction
Endometrial stromal sarcomas (ESSs), originating from endometrial mesenchymal cells, are rare uterine malignancies that account for approximately 0.2% of uterine malignancies [1,2]. The 2020 World Health Organization tumor classification system acknowledges four categories within the endometrial stromal tumors: endometrial stromal nodule, low-grade ESS (LGESS), high-grade ESS (HGESS), and undifferentiated uterus sarcoma (UUS), among which LGESS and HGESS are histologically, genetically and clinically distinct from UUS [3]. As reported in a large study on uterine mesenchymal tumors in China, LGESS and HGESS account for 81.2% and 18.8% of all ESSs with definite grade, respectively [4]. Despite the low incidence, HGESS patients have earlier and more frequent recurrences (typically within a year), and a higher risk of death than patients with LGESS [5]. Morphological and immunohistochemical findings are commonly used to distinguish HGESS and LGESS. Morphologically, LGESS tumor cells are small, oval to fusiform, and have low mitotic activity (usually less than five mitotic figures per 10 high-power fields [HPFs]). Most of the HGESS tumor cells were round with mitotic index ≥ 10 HPFs [6]. LGESS cells are usually positive for estrogen receptor (ER)/progesterone receptor (PR) and HGESS tumor cells often express cyclin D1 [7]. However, the morphological features and immunohistochemical markers used at present are not completely specific, and HGESS and LGESS cannot be completely distinguished using these characteristics [6].
Recently, genomic characterization has become an important tool for differential diagnosis of ESS. HGESS and LGESS frequently have distinct cytogenetic aberrations and specific chromosome translocations [8,9]. Tumors showing classic LGESS histology frequently contain genetic fusions between JAZF1 and members of polycomb complex genes (SUZ12, PHF1, and EPC1) [8]. In contrast, HGESSs are characterized by t(10;17) (q22;p13) chromosomal translocation, which results in YWHAE-NUTM2 fusion [10]. Other genetic alterations have also been reported in HGESS, including ZC3H7B-BCOR fusion and BCOR internal tandem duplication [9,11]. The unique chromosome translocations in HGESS and LGESS provide the molecular basis for accurate diagnosis and classification of ESS, and thus provide guidance for clinical management.
Currently, surgical resection (total hysterectomy±bilateral salpingo-oophorectomy) is the standard treatment for ESS. Besides, other therapies including chemotherapy, radiotherapy, and endocrine therapy can be considered [1]. As an emerging and promising treatment, immune checkpoint inhibitors and targeted drugs have significantly prolonged survival in other cancer patients (lung cancer, colorectal cancer, breast cancer, etc.). However, there have been few studies on their efficacy and efficacy predictors in ESS due to the rarity of this disease.
In this study, we comparatively analyzed the genomic and transcriptomic characteristics of 36 ESS patients from the perspective of individualized treatment, and explored the feasibility of targeted agents and immunotherapy for HGESS patients, which could provide the basis for the future design of clinical trials on precision treatment and personalized clinical management.
Materials and Methods
1. Patients and samples
This study retrospectively analyzed the sequencing data of 36 patients with a definite pathologic diagnosis of ESS who underwent genetic testing using next-generation sequencing (NGS) technology in Geneplus-Beijing (Beijing, China) from December 2019 to July 2022. The sample types included peripheral blood, formalin-immersed paraffin-embedded (FFPE) tissue, formalin-immersed tissue, and pleural effusion. All procedures were conducted in accordance with the Declaration of Helsinki. Samples were collected from patients after they had provided written informed consent. All experimental protocols were approved by Ethics Committee of Peking University People’s Hospital (Beijing, China) (approval No. 2022PHB419-001).
2. DNA-level NGS
Sample processing, DNA extraction, library preparation, hybridization and wash, sequencing, and bioinformatics analysis were performed as previously described [12–14]. Briefly, genomic DNA (gDNA) was extracted from the peripheral blood, FFPE, and pleural effusion samples using the CWE9600 Blood DNA kit (CoWin Biosciences, Cambridge, MA) and the Maxwell 16 FFPE Plus LEV DNA Purification kit (Promega, Madison, WI) according to the manufacturer’s instructions. The sequencing libraries of gDNA were constructed with the KAPA DNA Library Preparation Kit (Kapa Biosystems, Wilmington, MA). DNA libraries were then hybridized to a custom-designed biotinylated oligonucleotide probe covering 188–19,660 cancer-related genes (Integrated DNA Technology, Coralville, IA). DNA sequencing was then performed using the NextSeq CN 500 system (Illumina Inc, San Diego, CA), according to the manufacturer’s instructions.
The somatic single nucleotide variants (SNVs), small insertions and deletions (InDels), copy number variations (CNVs), and structural variations (SVs) were identified using the MuTect (v1.1.4), GATK (v3.4-46-gbc02625), Contra (v2. 0.8), and NoahCare structural variations detection (in house), respectively. Tumor mutation burden (TMB) was determined as the number of non-synonymous mutations per megabase and TMB-high (TMB-H) was defined as ≥ 10 muts/Mb [13]. Microsatellite instability (MSI) was evaluated using the MSIsensor (v0.5) software as previously described [14,15]. The threshold of MSI-high (MSI-H) was MSI score greater than 8%, which has been internally validated in 160 patients with multiple types of cancers, with both sensitivity and specificity of 100% compared with polymerase chain reaction (data not shown). Samples that did not meet the evaluation criteria of TMB and MSI were marked as TMB-unknown and MSI-unknown, respectively.
3. RNA sequencing
For 21 patients with tumor tissue available, RNA was extracted from the formalin fixation and paraffin embedding (FFPE) tumor samples using RNeasy FFPE Kit (QIAGEN, Hilden, Germany). RNA concentration, purity, and integrity were evaluated using the fluorometer (Qubit 2.0, Invitrogen, Carlsbad, CA), spectrophotometer (NanoPhotometer P330, Implen GmbH, München, Germany), and Bioanalyzer 2100 and the Agilent RNA 6000 Nano Kit (Agilent Technologies, Santa Clara, CA), respectively. Whole transcriptome sequencing was used to detect known and novel fusion genes (Geneplus-Beijing, Beijing). The mRNA with polyA tail was enriched by Oligo(dT) magnetic beads and then the fragmented mRNA was reversely transcribed into double-stranded cDNA using random primers, whose ends were modified, with A-tailing adenylated and adapters containing unique molecular tags and sample indexes ligated. Qualified libraries were sequenced using Geneplus 2000 sequencer (Geneplus-Beijing, Beijing). Bioinformatics analysis was performed using the inhouse software (forna V1.0.1). A gene fusion was determined when at least five high-quality unique reads spanned the breakpoint and minimum of three reads had a unique start site.
4. Programmed death-ligand 1 expression
The expression level of programmed death-ligand 1 (PD-L1) in tumor tissues was detected by monoclonal antibody 22C3 (Agilent Technologies) using Link 48 platform. The evaluation indexes included tumor proportion score (TPS) and combined positive score (CPS). TPS refers to the percentage of PD-L1 partial or total membrane staining positive tumor cells in the total tumor cells at any intensity. CPS refers to the ratio of PD-L1 stained cells (tumor cells, lymphocytes, and macrophages) in the total number of tumor cells multiplied by 100. TPS ≥ 1% and/or CPS ≥ 1 were defined as positive for tumor PD-L1 expression [16].
5. Public data acquisition and data analysis
To investigate the most frequently mutated genes and pathways in ESS, the mutation data of 64 ESS patients from four public datasets was downloaded from cBioPortal (https://www.cbioportal.org/) and compared to our cohort (pmid: 33106634, 28481359, 35705560, and 32299819). The samples were sequenced using one of three versions of MSK-IMPACT panel covering 341, 410, or 468 genes. The races of the patients included in MSK dataset included white, black, Asian, and others.
R package ggvenn 0.1.9 was used for finding the overlapping genes between our dataset and MSK dataset. R package clusterProfiler 4.4.4 was applied to perform the Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis. Differentially expressed gene analysis between HGESS and LGESS was performed using the DESeq2 1.36.0 package, and volcano plot was drawn using the ggpubr 0.4.0 package and ggthemes 4.2.4 package. R package GSVA 1.44.3 was used for single sample Gene Set Enrichment Analysis (ssGSEA) and the marker genes of immune cell types for ssGSEA were obtained from the previous literature [17].
Student t test and Fisher’s exact test were used to access continuous and categorical variables between HGESS and LGESS, respectively. Differences in ages at diagnosis between HGESS and LGESS were examined with the Mann-Whitney test. Differences in operation history, drug history, radiotherapy history, metastasis, and the expression of ER and PR were examined with two-sided Fisher’s exact test. p < 0.05 was considered statistically significant. Statistical analyses were performed using GraphPad Prism (9.0.0, GraphPad Software Inc., San Diego, CA).
Results
1. Clinicopathologic characteristics
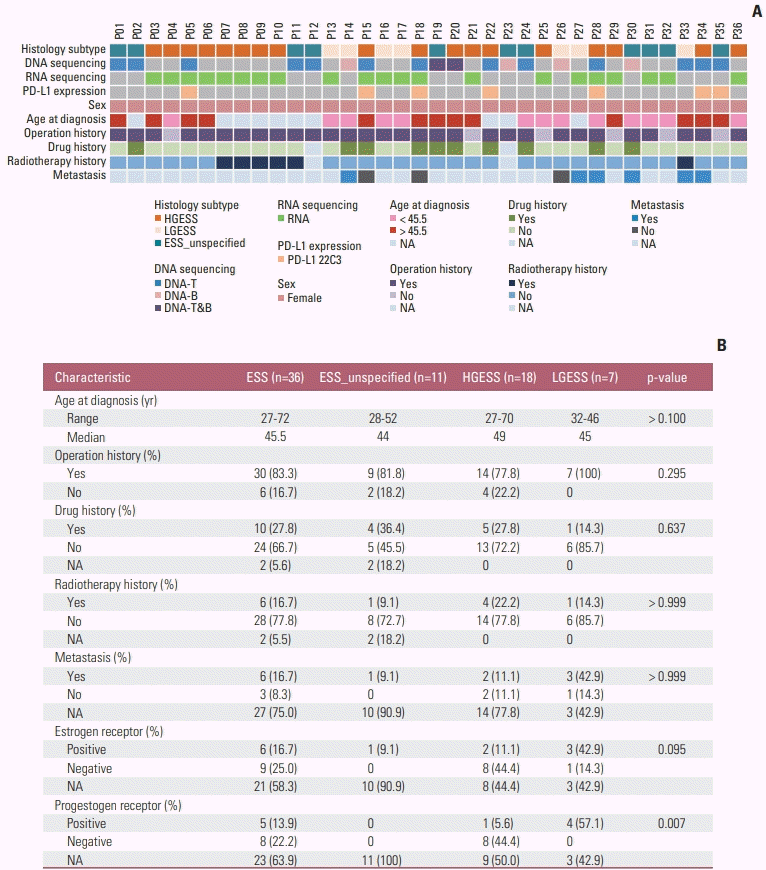
The baseline characteristics of patients in our cohort were summarized in Fig. 1. A total of 36 patients were enrolled in this study, including 18 HGESS, 7 LGESS, and 11 ESS patients with unspecified grade (Fig. 1A). Of the 36 patients, 19 patients underwent DNA sequencing (P35: whole exome sequencing; others: panel sequencing), 21 patients underwent RNA sequencing, and seven patients underwent PD-L1 testing. The panel used for DNA sequencing covered 188–1,021 genes. The most commonly used panel covered 1,021 genes, accounting for 63.2% of DNA sequencing.
All patients were female and the age at diagnosis ranged from 27 to 72 years (median age, 45.5 years). Of the 36 patients, 83.3% had a history of surgery, 27.8% had received drug therapy, 16.7% had received radiotherapy, and 16.7% had tumor metastasis (Fig. 1A). There was no significant difference between HGESS and LGESS patients in terms of the age at diagnosis, operation history, drug history, radiotherapy history, metastasis and ER expression (p > 0.05). There was a highly significant difference between HGESS and LGESS in PR expression (HGESS vs. LGESS, 10% vs. 100%; p < 0.01) (Fig. 1B).
2. Mutational spectrum of ESS in our and MSK cohorts
A total of 198 mutations were detected in 28 of 36 patients in our cohort, including 80 SNVs, 61 CNVs, 41 SVs, and 16 InDels. Of the eight patients without detectable mutations, five underwent RNA testing (P04, P06, P16, P21, and P29) and three patients (P19, P26, and P30) underwent DNA sequencing covered 365, 386, and 1,021 genes, respectively. A total of 169 somatic mutations were detected in 64 patients in MSK cohort, including 70 CNVs, 73 SNVs, 10 SVs, 16 InDels. Mutations that occur more than twice were present in Fig. 2. In our cohort, we found that the most frequently mutated gene was TP53 (31.6%). ZC3H7B-BCOR fusion was the most frequent mutation in HGESS (40%). Two patients in HGESS detected NUTM2B-YWHAE fusion (13.3%). All mutations occur only once in LGESS patients (Fig. 2A). In MSK cohort, DNMT3A was the most common mutation (28.1%). JAZF1-SUZ12 was the most frequent mutation in LGESS (27.3%) (Fig. 2B). NUTM2B-YWHAE fusion occurred in both HGESS and LGESS, with a percentage of 9.1%.
As shown in Fig. 2C, there are 26 commonly mutated genes in both cohorts. These intersected genes were significantly enriched in phosphoinositide 3-kinase (PI3K)–Akt signaling pathway, cellular senescence, cell cycle, P53 signaling pathway, endocrine resistance, and other pathways. The genes related to endocrine resistance pathway include NOTCH3, CDKN2A, CDK4, MDM2, MAPK1, TP53, and MTOR. In GenePlus cohort, a total of 12 patients had endocrine resistance pathway-related mutations, including 6 HGESS patients (P05, P15, P18, P20, P22, and P34) and 1 LGESS patient (P14). Five of the seven patients underwent hormone receptors testing, with four of the HGESS patients negative for ER and PR (P05, P18, P20 and P34). ER positivity was detected in only one patient (P15) with copy number loss of CDKN2A detected.
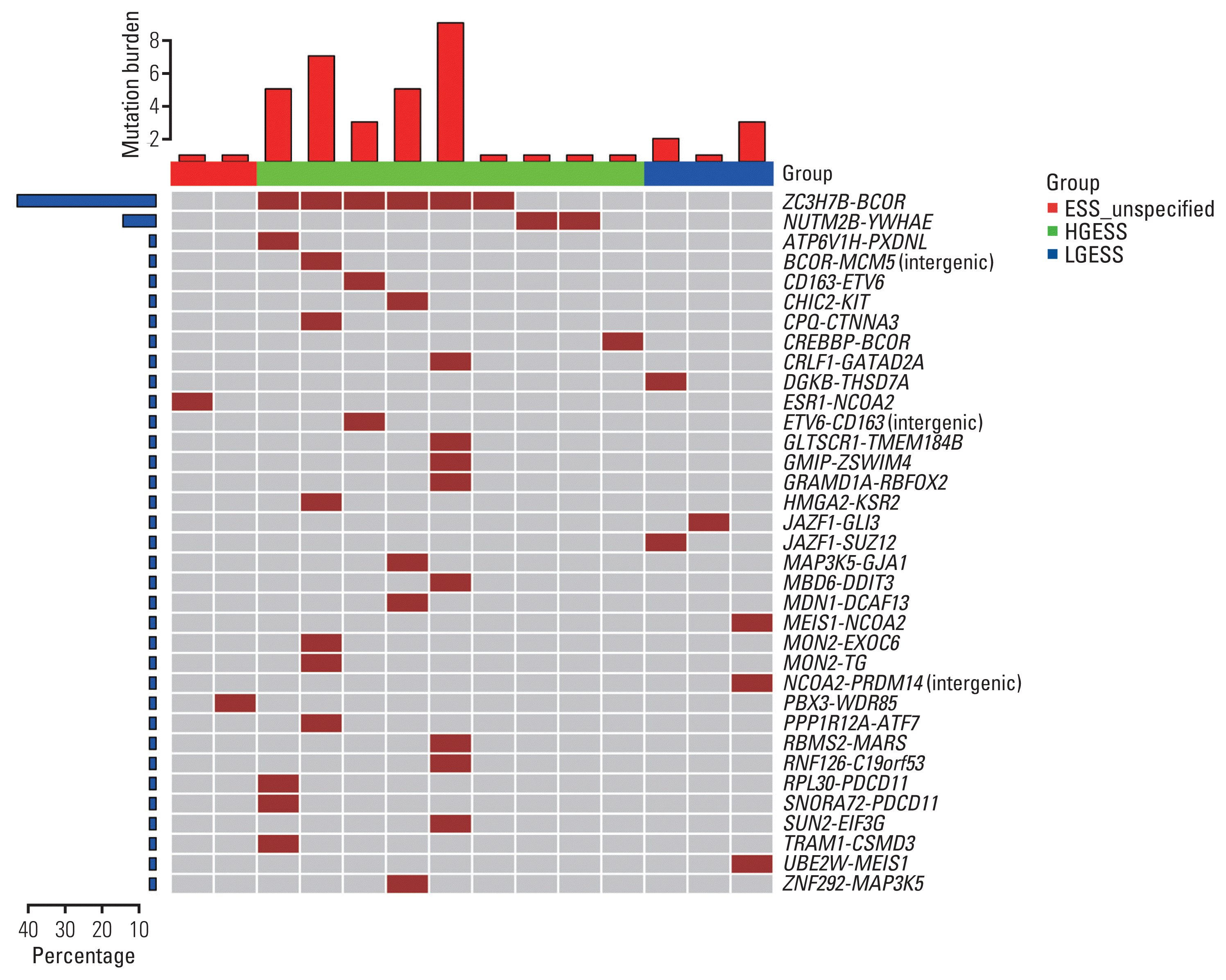
3. Fusions identified through RNA sequencing in 21 ESS patients
A total of 21 patients in our cohort underwent RNA sequencing. A total of 35 fusions were detected in 14 patients, with the fusion detection rate of 66.7%. As shown in Fig. 3, the most common fusion in our cohort was ZC3H7B-BCOR (28.6%). In HGESS group, the most frequent fusions were ZC3H7B-BCOR (40%) and NUTM2B-YWHAE (13.3%). In LGESS group, the most frequent fusions were JAZF1-related fusions, with partner genes of SUZ12 (25%) and GLI3 (25%). In ESS, classical fusions include ZC3H7B-BCOR fusion and NUTM2A/B-YWHAE fusion, which are representative in HGESS, and JAZF1-SUZ12 fusion, which is frequently found in LGESS. The classical fusion detection rate in this cohort was 42.9%. The detection rate of classical fusion was 53.3% in HGESS patients and 25.0% in LGESS patients.
In addition to classical fusions, some non-classical fusions that are not frequently found in ESS were also detected. For example, CREBBP-BCOR was detected in one HGESS patient and MEIS1-NCOA2 was detected in one LGESS patient. Two non-classical fusions, ESR1-NCOA2 and PBX3-WDR85, were detected in ESS patients with tumor grade unspecified.
4. RNA expression profiles between HGESS and LGESS of our cohorts
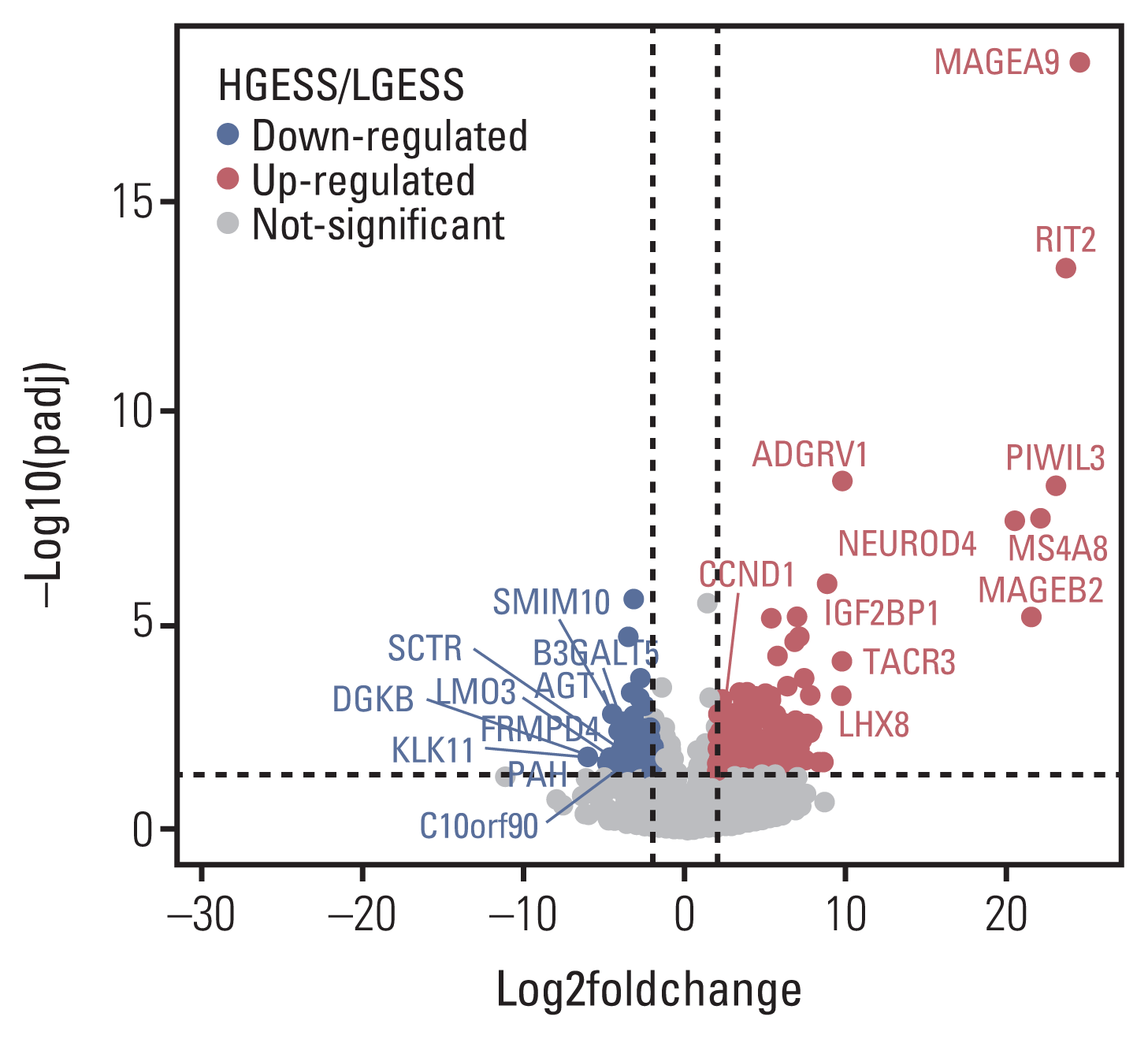
In order to analyze the differences between HGESS and LGESS from the perspective of RNA expression, we selected 15 patients with HGESS and four patients with LGESS from a total of 21 patients who underwent RNA sequencing. Differential expression analysis between HGESS and LGESS groups was performed. The results were shown in Fig. 4. A total of 298 genes were significantly differentially expressed between HGESS and LGESS by greater than 4-fold with 5% false discovery rate (FDR) cutoff with Benjamini-Hochberg correction, of which 234 and 64 genes were overexpressed in HGESS and LGESS, respectively. The top10 genes overexpressed in HGESS included MAGEA9, RIT2, PIWIL3, MS4A8, MAGEB2, NEUROD4, TACR3, ADGRV1, LHX8, and IGF2BP1. The top10 genes overexpressed in LGESS included KLK11, DGKB, FRMPD4, SMIM10, C10orf90, AGT, PAH, SCTR, B3GALT5, and LMO3. In addition, CCND1 gene was one of the genes that significantly up-regulated in HGESS (Log2FC=2.10 and FDR=0.03). The expression of GPER1 and PGR that encode ER and PR were not significantly different between HGESS and LGESS.
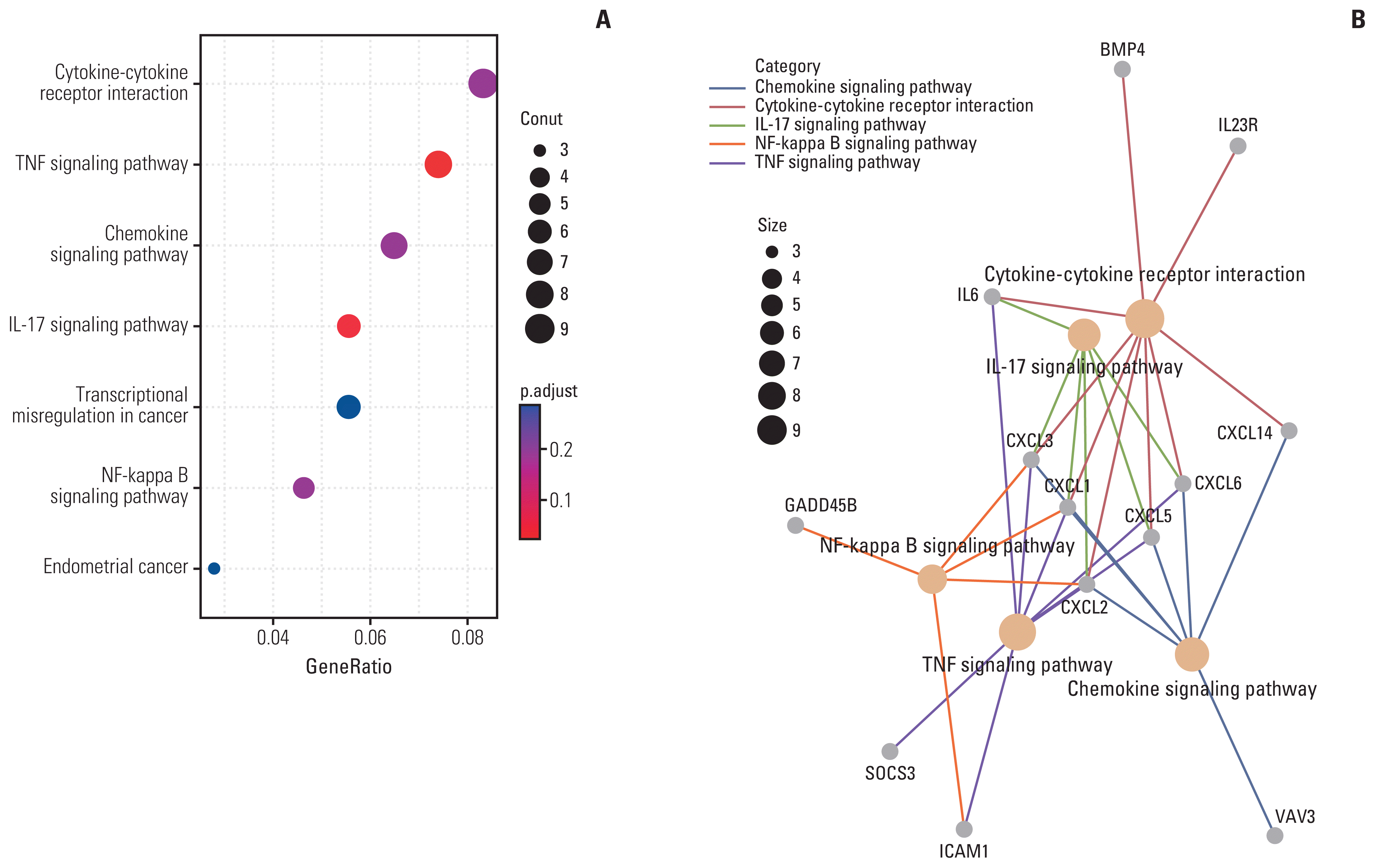
KEGG enrichment analysis of significantly up-regulated genes in the HGESS and LGESS groups, respectively, showed that there were significantly enriched pathways in both groups (p < 0.05). In HGESS group, up-regulated genes were significantly enriched in pathways including cytokine-cytokine receptor interaction, tumor necrosis factor signaling pathway, chemokine signaling pathway, interleukin-17 signaling pathway, transcriptional misregulation in cancer, nuclear factor κB signaling pathway, endometrial cancer, and so on (Fig. 5A). Most of these pathways was associated with immunity. As shown in Fig. 5B, the intersecting genes in all five immune-related pathways belonged to the chemokine family. In LGESS group, up-related genes were significantly enriched in PI3K-Akt signaling pathway.
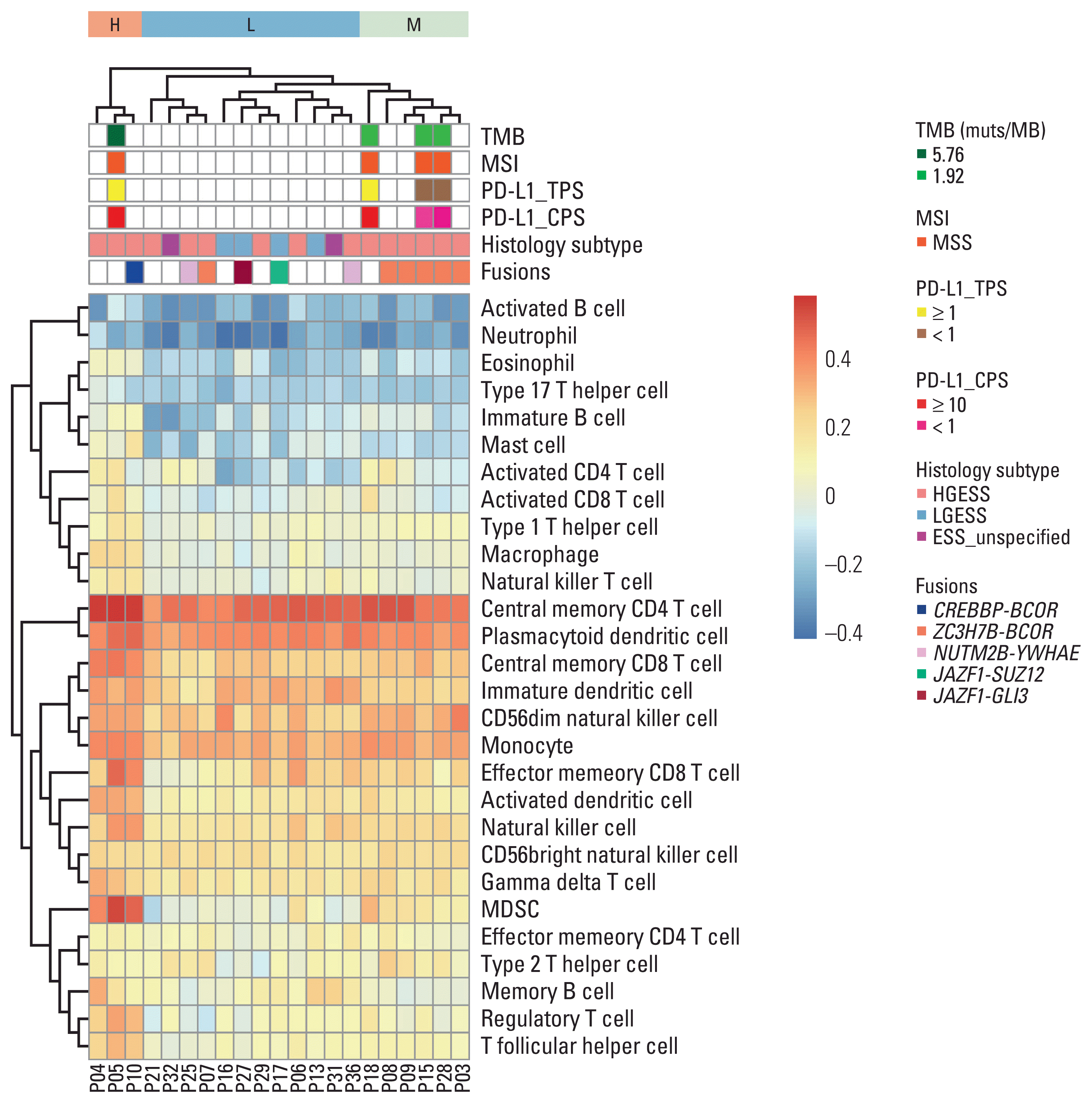
5. Tumor-immune microenvironment of 21 ESS patients
The transcriptomic profiles of 21 patients in our cohort were used to analyze immune infiltration. Four of them were evaluated for TMB, MSI, and PD-L1 (P5, P15, P18, and P28). All four patients were microsatellite stable and TMB-low. Two of the four patients were positive for PD-L1, and the remaining two were negative for PD-L1.
Based on the results of hierarchical unsupervised clustering, we divided these 21 patients into three groups (Fig. 6). Group H had the highest degree of immune infiltration (n=3), and the disease classification of them was all HGESS (P04, P05, and P10). The level of immune infiltration was lowest in group L (n=12). All LGESS cases belonged to group L. The proportion of patients with HGESS and LGESS in the L group was 50% and 33.3%, respectively. Group M had a relatively moderate degree of immune infiltration (n=6) and all these patients were HGESS (P03, P08, P09, P15, P18, and P28). Group H was characterized by the highest infiltration of central memory CD4 T cell, myeloid-derived suppressor cell (MDSC), central memory CD8 T cell, plasmacytoid dendritic cell, effector memory CD8 T cell, regulatory T cell, and T follicular helper cell. Notably, the P05 patients in group H had positive PD-L1 expression and the highest TMB value. Patients in group M exhibited a relatively moderate level of immune infiltration of central memory CD4 T cell, CD56dim natural killer cell, and MDSC. In addition, P18 patients in this group also presented with positive expression of PD-L1. The degree of immune infiltration in group L was opposite to that in the H and M groups, showing the characteristics of relatively low immune infiltration of central memory CD4 T cell, neutrophil, activated CD8 T cell, MDSC cell, and regulatory T cell.
The association between fusions and immune infiltration was also investigated. CREBBP-BCOR fusion was detected in one of three patients (HGESS) in group H. ZC3H7B-BCOR fusion was seen in five of the six patients (HGESS) in group M. Among the 12 patients in group L, ZC3H7B-BCOR (HGESS) was detected in one patient, NUTM2B-YWHAE was detected in two patients (HGESS), and JAZF1-related fusion was detected in two patients (LGESS, fusion partners were SUZ12 and GLI3, respectively).
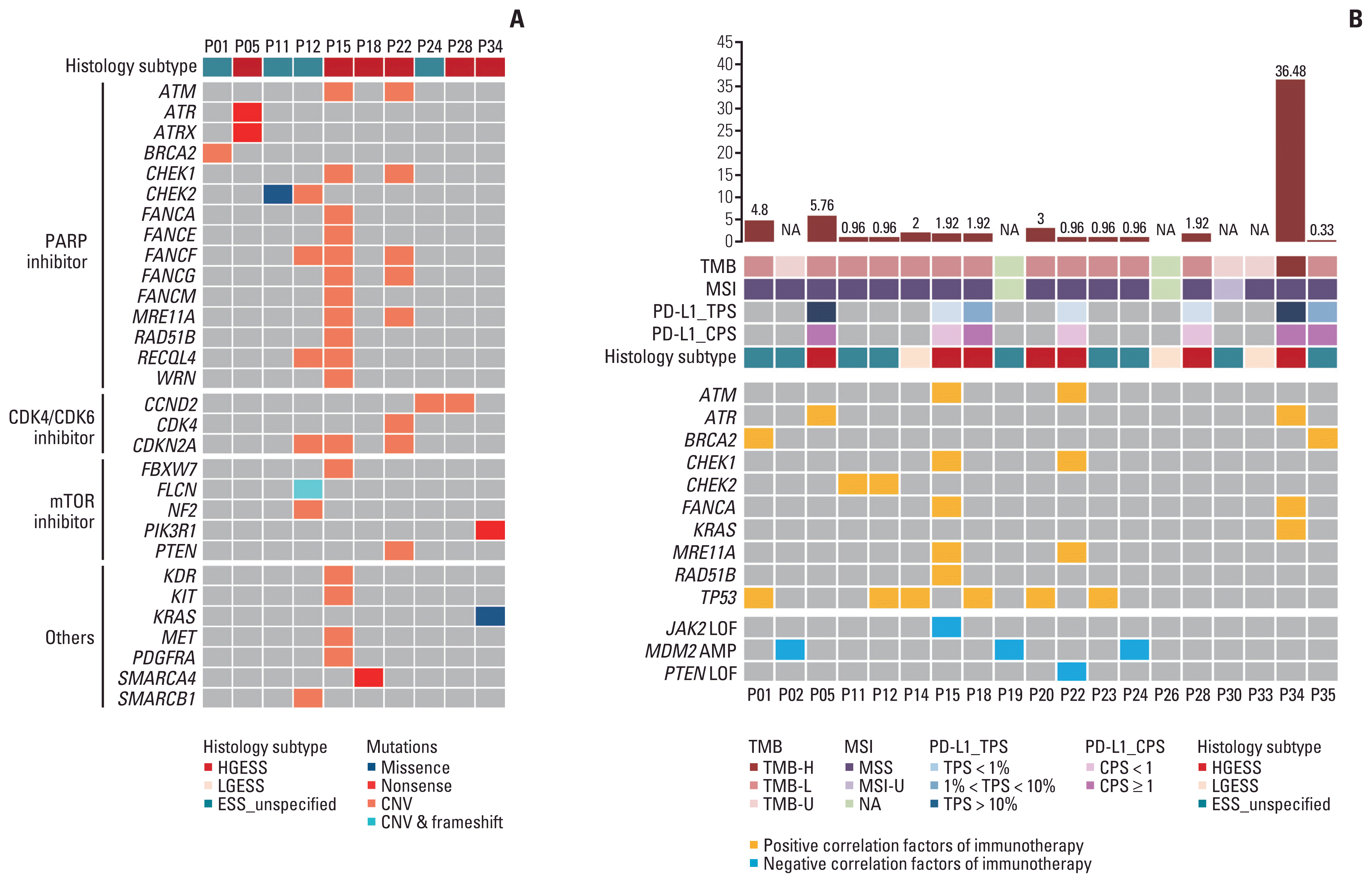
6. Actionable mutations and immunotherapy-related biomarkers in our cohort
Next, the possibility of targeted therapy and immunotherapy for ESS patients was explored (Fig. 7). In our cohort, targeted drug-associated mutations were detected in 10 of 36 patients, with a detection rate of 27.8%. Of these 10 patients, 60% (n=6) patients had HGESS, and the remaining patients had unspecified tumor grade. Based on the types of targeted inhibitors indicated as sensitive, we divided these genes into three main groups (poly ADP-ribose polymerase [PARP] inhibitor, CDK4/6 inhibitor, and mammalian target of rapamycin [mTOR] inhibitor groups) (Fig. 7A). The three groups were enriched in homologous recombination repair process, cell cycle signaling, and PI3K/AKT/mTOR signaling pathways, respectively, which indicated that PARP, CDK4/6, and mTOR inhibitors may be applied to patients with specific gene mutations. Six out of the 10 patients were sensitive to PARP inhibitors, and 50.0% of them were HGESS patients (n=3). Five of the 10 patients were sensitive to CDK4/CDK6 inhibitors, and 60% of them were HGESS patients (n=3). Four of the 10 patients were sensitive to mTOR inhibitors, and 75.0% of them were HGESS patients (n=3).
The genomic biomarkers of immunotherapy efficacy were also evaluated in ESS patients who underwent DNA sequencing (Fig. 7B). The detection rate of TMB-H, MSI-H, and PD-L1 expression was 5.3%, 0, and 57.1%, respectively. Of these 19 patients, 36.8% (n=7) belonged to HGESS, of which four patients had positive immunotherapy efficacy–related mutations and two patients had both positive and negative immunotherapy efficacy–related mutations. Fifteen point eight percent (n=3) of the patients belonged to LGESS, of which one patient was detected with positive immunotherapy efficacy–related indicators. On the whole, positive and negative indicators of immunotherapy efficacy were detected in 12 and five patients, respectively. In addition to KRAS and TP53 mutations, positive immunotherapy efficacy–related features were found in the DNA damage repair pathway. A total of three negative correlation genes of immunotherapy efficacy were detected in our cohort, including JAK2 loss-of-function (LOF) mutations, MDM2 amplification, and PTEN LOF mutations. The detection of these three biomarkers implied a low benefit of immunotherapy in patients with P02, P15, P19, P22, and P24.
Discussion
As a rare malignancy, the diagnosis of ESS is challenging, and patients with ESS (especially HGESS) have limited options for treatment. Our study attempted to expand the potential therapeutic options for patients with HGESS by analyzing and comparing the characteristics of HGESS and LGESS patients at the genomic and transcriptomic levels. This study significantly increases our understanding of molecular characteristics of HGESS and LGESS, and suggests that targeted therapy and immunotherapy may be feasible in HGESS patients with specific molecular features.
This study found that CNVs and SVs frequently occurred in patients with ESS, and TP53 and DNMT3A gene mutations were the most common mutations (31.6% and 28.1%). TP53 mutation was frequently detected in other uterine sarcomas such as uterine leiomyosarcoma and UUS, while DNM-T3A mutation was more common in myeloid malignancies [18–20]. To our knowledge, this is the first study to definitely report the frequent occurrence of these two mutations in ESS.
Consistent with previous literature [8,9], the classical fusions frequently found in HGESS (ZC3H7B-BCOR and NUTM2B-YWHAE) and LGESS (JAZF1-SUZ12) were detected in our cohort. The detection rate of classical fusion in our study was 42.9%, which was slightly higher than that reported by Juckett et al. [21] using the FMI database (28.7%). This occurs probably due to the fact that the panel used in the study did not include the YWHAE fusion. Although the fusions found in HGESS and LGESS are specific, there are occasional exceptions. It is worth noting that in MSK cohort of our study, one patient with NUTM2B-YWHAE fusion was diagnosed with LGESS. One study also reported a case of ESS with complete LGESS morphology, strong and diffuse expression of ER and PR, and detection of YWHAE-NUTM2MA/B fusion [22]. This incidental case reminds us that the diagnosis and clinical management of ESS patients should be based not only on molecular subtyping results but also on clinicopathological features.
In addition to classical fusion, non-classical fusions were often detected in ESS. With the advancement of NGS technology, the classification of uterine sarcoma is constantly developing, and new molecular features are constantly being incorporated. In our study, CREBBP-BCOR fusion was detected in a patient with HGESS, while this fusion had been detected as a novel BCOR gene fusion in uterine sarcoma in a previous study [11]. In addition, ESRI-NCOA2 fusion with tumor grade unspecified in our cohort was also detected in uterine tumors resembling ovarian sex cord tumor [23]. For the non-classical fusions detected in ESS, more samples need to be collected to effectively analyze the relationship between genetic characteristics and disease classification and prognosis.
We identified genes with up-regulated expression in HGESS and LGESS. Notably, overexpression of MAGEA9 gene was associated with poor prognosis in uterine cancers [24], which may contribute to the aggressive behavior of HGESS. Up-regulated expression of MAGEB2 gene promoted laryngeal cancer cell invasion and correlated with favorable immunotherapy efficiency [25]. The protein encoded by the PIWI3 gene was associated with metastasis in advanced epithelial ovarian cancer [26]. Overexpression of IGF2BP1 promoted the progression of endometrial cancer [27].
In clinics, expression of cyclin D1, ER, and PR are often used to assist in ESS classification [7]. This study showed that the expression of the CCND1 gene was significantly up-regulated in HGESS, which verified the value of the cyclin D1 protein encoded by the CCND1 gene in the diagnosis of HGESS. Nevertheless, the expression of GPER1 and PGR genes encoding ER and PR did not differ significantly between HGESS and LGESS, which showed the limitation of these two markers. Besides, the classical fusion in HGESS and LGESS is not completely specific, and molecular characteristics cannot be used as the sole indicator for accurate classification of ESS. This suggests that morphological, pathological, and molecular features should be combined to making the clinical diagnosis of ESS.
The possibility of targeted therapy and immunotherapy in ESS patients was explored in our study. In GenePlus cohort, 60% of the patients with targeted drug-related mutations were in HGESS, and no targeted drug-related mutations were detected in LGESS. Patients with detectable targeted drug-related mutations may be sensitive to PARP, mTOR, and CDK4/6 inhibitors. The common mutations in GenePlus and MSK cohorts in our study were significantly enriched in PI3K-Akt signaling, cellular senescence, cell cycle, P53 signaling, and endocrine resistance pathways. In GenePlus cohort, more HGESS patients had mutations associated with the endocrine resistance pathway compared to LGESS patients, and the vast majority of HGESS patients had negative hormone receptor results. The lack of ER and PR expression and the presence of endocrine resistance-associated mutations may lead to primary drug resistance of endocrine therapy in HGESS patients, even in HGESS patients with positive ER receptor expression. This is consistent with the finding that patients with HGESS are not suitable for endocrine therapy [1]. Although no targeted drug-related mutations were detected in LGESS in our cohort, pathway enrichment analysis of up-regulated genes in LGESS revealed significant enrichment of PI3K-Akt signaling pathway, suggesting that PI3K or mTOR inhibitors could be considered in these patients. A study showed the clinical benefits of mTOR inhibitors in four patients with both TSC2 mutation and JAZF1-SUZ12 fusion [28].
In our study, up-regulated genes in HGESS were significantly enriched in five immune-related pathways. In addition, most HGESS patients (85.7%) had positive genomic biomarkers of immunotherapy efficacy, while only 33.3% of LGESS patients had positive factors. Moreover, the results of immune microenvironment analysis showed that both the H and M groups with relatively high immune infiltration were HGESS patients, and two of them had PD-L1 expression. By studying the tumor microenvironment in HGESS, Kim et al. [29] concluded that immune checkpoint inhibitors may be excellent candidates for future treatment of HGESS. In addition, an analysis of immune infiltration in ESS patients showed that two HGESS patients with YWHAE-NUTM2 fusion had a very low degree of immune infiltration [30]. In our research, the degree of immune infiltration in patients with ZC3H7B-BCOR fusion HGESS was relatively higher than that in patients with NUTM2B-YWHAE, implying that the degree of immune infiltration of HGESS may be related to different fusions. Overall, we proposed that HGESS has an immune-hot microenvironment. HGESS patients with PD-L1 positivity, deeper immune infiltration, positive genomic indicators, and/or upregulation of gene expression in immune-related pathways may be suitable for immunotherapy, but also need to be alert to drug resistance and negative genomic indicators.
Due to the rarity of ESS, several potential limitations of this study need to be mentioned. Firstly, due to the limited number of LGESS patients in our cohort, differential expression analysis results may lack precision and mutational profiles of LGESS may not be representative. Secondly, the number of patients undergoing PD-L1 detection in the cohort is small, making it nearly impossible to accurately combine immune infiltration characteristics and PD-L1 results in the analysis of tumor-immune microenvironment. Large-scale studies should be conducted in the future to validate the findings of our studies.
In conclusion, our study describes the molecular characteristics of ESS and its subtypes at the genomic and transcriptomic levels, addresses the differences in molecular characteristics between HGESS and LGESS, and explores the potential feasibility of targeted therapy and immunotherapy in patients with HGESS. Accurate diagnosis of ESS requires a combination of morphological, pathological, and molecular features. Potential treatment options for HGESS include targeted therapy and immunotherapy, and actionable mutations, PD-L1 expression, immune microenvironment characteristics, and immunotherapy-related genomic biomarkers should be evaluated to guide the decision-making of treatment. NGS is a powerful tool to support accurate diagnosis and clinical precision drug administration, making personalized clinical management of ESS patients possible.

 XML Download
XML Download