

 PDF
PDF Citation
Citation Print
Print



Introduction
Lung cancer remains the leading cause of cancer-related deaths in both men and women [1]. Recently, the use of targeted therapy for driver mutation-positive lung cancer and the implementation of immune checkpoint inhibitors have improved survival rates [2,3]. However, targetable driver mutations cannot be identified in approximately 40% of lung cancer patients, and no effective treatment exists for patients in whom immunotherapy fails [4,5].
Uncontrolled growth, proliferation, and survival of cancer occurs through the mammalian target of rapamycin (mTOR) signaling pathway in patients with driver mutations in genes such as EGFR, ALK, ROS, or cMET, as well as in lung cancer patients without effective target agents. Thus, drugs targeting the mTOR pathway can be used to block cancer growth [6]. Rapamycin is the representative first-generation mTOR inhibitor. It binds to the immunophilin FKBP12 and inhibits some functions of mTOR complex 1 (mTORC1). Second-generation mTOR inhibitors are designed to act as ATP-competitors of mTOR and counteract mTOR activity at half-maximal inhibitory concentrations (IC50) less than that of phosphoinositide 3-kinase (PI3K) [7]. However, these mTOR inhibitors can promote survival and metastasis of cancer cells through a feedback pathway, and can also harm normal tissues [8].
Leucyl-tRNA synthetase 1 (LARS1) functions as a GTPase-activating protein by binding to Ras-related GTP-binding protein (Rag) GTPase in a manner that is dependent on the leucine concentration in the cytoplasm. This implies that mTORC1 activity could be reduced by inhibiting the binding of LARS1 to Rag GTPase and further suggests that tumor growth could be inhibited by targeting this pathway [9,10]. We previously showed that BC-LI-0186 binds to the RagD interacting site of LARS1, thus inhibiting lysosomal localization of LARS1 and mTORC1 activity [2,9]. However, due to the bypass activity of the uninhibited mitogen-activated protein kinase (MAPK) pathway, which is located above the site of action of BC-LI-0186, cancers may continue to progress [2].
Trametinib is approved by the U.S. Food and Drug Administration for BRAF V600E mutation-positive non–small cell lung cancer (NSCLC) and targets MEK1/2 in the MAPK pathway [11]. We hypothesised that, if BC-LI-0186 and trametinib were used simultaneously for lung cancer without identifiable mutations, the therapeutic effect may be enhanced.
We thus aimed to investigate the cause of limited efficacy of BC-L1-0186 and the combined effects of BC-LI-0186 and trametinib on NSCLC in cell culture and xenograft models, to shed light on the cause of the limited efficacy of BC-LI-0186.
Materials and Methods
1. Cell lines, chemicals, and antibodies
The cell lines used in this study were purchased from the Korean Cell Line Bank (https://cellbank.snu.ac.kr/main/; Seoul, Korea). BC-LI-0186 and Trametinib were purchased from Selleckchem (cat No. E0488, Houston, TX). Anti-LARS1 antibody was purchased from Neomix (Suwon, Korea) and the others were described in the S1 Table.
2. Immunoblotting
Cells were harvested on ice using 2× LSL buffer, containing Xpert Duo Inhibitor Cocktail Solution (GenDEPOT, Baker, TX). After sonication, 30–50 μg of lysate was separated by electrophoresis on 7.5%–12.0% polyacrylamide gels, after which proteins were transferred to nitrocellulose membranes (Bio-Rad Laboratories, Inc., Richmond, CA). Each protein’s expression level was measured using ImageJ software (http://rsbweb.nih.gov/ij/) and quantified relative to that of β-actin [12].
3. Differentially expressed genes
To identify differentially expressed genes (DEGs), RNA sequencing of the BC-LI-0186-sensitive cell line, H460, and -resistant cell line, A549, was performed by Macrogen (Seoul, Korea). RNA sequencing data were obtained from three independent experiments, using pairs of cell samples cultured under the same conditions. Raw count data were normalised by the median ratio method in the size of patient samples and were transformed using the regularised log-transformation function in DESeq2 packages (ver. 3.15, https://bioconductor.org/packages/release/bioc/html/DESeq2.html).
4. Cell death, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenylte-trazolium bromide assay and drug combination study
To measure cell death, cells were treated with the indicated dose of BC-LI-0186 for 48 hours, then stained with annexin V and propidium iodide (PI) and analyzed using a FACSCanto II flow cytometer (Becton Dickinson, Franklin Lakes, NJ). The effect of the treatment on cell proliferation was assessed using a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Briefly, 5×105 cells/well were treated with BC-LI-0186 (0–16 μM), trametinib (0–96 nM), or their combination at fixed BC-LI-0186:trametinib concentration ratios of 1:0.5, 1:1, and 1:2. After 48 hours, MTT was added at a final concentration of 0.5 mg/mL, and cells were incubated for an additional 2 hours at 37°C. Formazan complexes were dissolved in dimethyl sulfoxide, and the 550-nm absorbance was measured spectrophotometrically (Thermo Scientific, Rockford, IL). The combination index (CI), a quantitative measure of the interaction between drugs, was determined. A CI > 1 indicates a synergistic effect, CI=1 indicates an addictive effect, and CI < 1 indicates an antagonistic effect [10].
5. Immunocytochemistry
A549 and H460 cells (5×105 cells/well) were plated in 6-well plates containing sterilised coverslips. The next day, cells were fixed with 4% formaldehyde in phosphate-buffered saline (PBS), incubated in blocking solution containing 5% bovine serum albumin, and then incubated with anti-LC3A/B rabbit polyclonal antibody (Abcam, Cambridge, MA) and anti-LAMP-2 antibody (ABL93). The next day, cells were washed and Alexa Fluor 488-conjugated anti-rabbit IgG secondary antibodies were added. Nuclei were counterstained with DAPI (1:1,000) and cells were imaged using an LMS 710 confocal microscope (Carl Zeiss, Oberkochen, Germany). Images were analyzed using ZEN imaging software (ver. 8.0.0.273, Carl Zeiss).
6. Animal experiment
Mouse experiments were approved by the Institutional Animal Care and Use Committee of the Yonsei Laboratory Animal Center (#2018-0242). Eighteen BALB/c nude mice, purchased from Orient Bio (Seongnam, Korea) were used; Vehicle group (n=4), Trametinib group (n=5), BC-LI-0186 group (n=4), and Combination group (n=5). H460 cells (1.0× 107) were injected subcutaneously into the flank of BALB/c nude mice at around 9 weeks of age. These mice were randomly assigned to four groups after stratification of the tumor volume (TV). Mice groups received the following treatment: vehicle (10% dimethylacetamide and 10% Tween 80 in PBS), BC-LI-0186 (20 mg/kg bid, 5 days/week, i.p.), trametinib (1 mg/kg, 5 days/week, oral), or a combination of both drugs for 2 weeks. The TV was calculated using the formula: V=1/2×(length×width2) [13,14].
7. Statistical analysis
Independent sample t tests were used for univariate analysis of continuous variables. Differences in TV and body weight among mouse groups were analyzed using the Mann-Whitney U test. SPSS ver. 26 (IBM Corp., Armonk, NY) and R (ver. 4.1.0, https://www.r-project.org/) software were used for statistical analyses, which were two-tailed. p < 0.05 were interpreted as indicating statistical significance.
Results
1. Positive correlation between LARS1 expression and mTORC1 activity
First, the relationship between the expression of LARS1 and pS6, a surrogate for mTORC1 activity, was evaluated using stable NSCLC cell line lysates. LARS1 activates the mTORC1 pathway by sensing leucine. In NSCLC cell lines maintained in growth media supplemented with 5% fetal bovine serum (FBS), the expression levels of LARS1 and pS6 were significantly positively correlated (p=0.004).
LARS1 expression also correlated positively with MEK phosphorylation (pMEK; p=0.013), but not with extracellular signal-regulated kinase (pERK) or AKT (pAKT) phosphorylation (Fig. 1A and B). H460 and A549 cells were treated with leucine in FBS (−) and leucine (−) media. Leucine treatment induced dose-dependent phosphorylation of S6 but did not affect MEK, ERK, or AKT phosphorylation. This suggests that mTORC1 activation by LARS1 is independent of the AKT or MAPK pathways (Fig. 1C). When BC-LI-0186, which selectively blocks LARS1 and RagD interaction by competing with RagD for LARS1 VC-domain binding, was used to treat lung cancer cells in FBS (−) media, decay of S6 phosphorylation was observed after 3 hours (Fig. 1D). Additionally, we evaluated the dose-dependent mTORC1 inhibition by BC-LI-0186 under the presence of growth factors. At a low dose of 1 μm or less, pAkt, pGSK3B, pp70S6K, p4EBP1, and pS6 expressions were elevated, and at a high dose of 10 μm or more, the expressions were decreased (Fig. 1E).
Fig. 1
Expression of leucyl-tRNA synthetase 1 (LARS1) correlates positively with the mammalian target of rapamycin complex 1 (mTORC1) signal in non–small cell lung cancer (NSCLC). (A) Immunoblotting of LARS1 and molecules involved in mTOR signaling in various NSCLC cells. (B) The correlation plot between LARS1 and pS6 (r=0.8156, p=0.0040), pAkt (r=0.5879, p=0.0739), pERK (r=0.4092, p=0.2403), and pMEK (r=0.7478, p=0.0129). There was a significant positive correlation between LARS1 and pS6. p-values were obtained from Pearson’s correlation analysis and r denotes Pearson’s correlation coefficient. (C) Immunoblotting and heatmap of LARS1 and molecules associated with mTOR signaling according to leucine concentration in the A549 cell line and H460 cell line. (D) Effects of BC-LI-0186 (10 μM) on the A549 and H460 cell lines over time in fetal bovine serum (FBS) (−) media. (E) Effects of the BC-LI-0186 concentration on the A549 and H460 cell lines in FBS (+) media. It was observed that BC-LI-0186 concentrations less than 10 μM had a paradoxical effect on both A549 and H460 cell lines. R*, each cell line was treated with rapamycin (10 μM) for 10 minutes and subjected immunoblotting; S**, serum shock (used for positive control).
![]()
Fig. 1D and E show relatively long time and a high dose of 10 μm or more are required for BC-LI-0186 to exhibit an effect on cancer cells. These results demonstrate that BC-LI-0186 alone cannot effectively inhibit cancer cells.
2. BC-LI-0186 paradoxically activates mTORC1 signaling and induces MEK/ERK phosphorylation in the presence of growth factors
To find the causes of paradoxical phenomenon, the effect of BC-LI-0186, was evaluated in the presence of growth factors, to mimic the in vivo environment better. S6 phosphorylation paradoxically increased with BC-LI-0186 treatment of lung cancer cells cultured in growth media containing 5% FBS. Additionally, MEK and ERK phosphorylation increased with increasing BC-LI-0186 (Fig. 2A). This suggested that activation of the reciprocal MAPK pathway reduces the therapeutic efficacy of BC-LI-0186 in cancer.
Fig. 2
Paradoxical response of BC-LI-0186 to lung cancer cell line in the presence of growth factors. (A) Immunoblotting and line graph of pMEK, pERK, and pS6 expression according to the concentration of BC-LI-0186 in the A549 and H460 cell lines. (B) H460 and A549 cell lines were treated with BC-LI-0186 (10 μM) or cisplatin (50 μM) for 48 hours, and cell death was measured by flow cytometry unsing annexin V and propidium iodide (PI) staining. (C) Volcano plot. The log2 fold-change indicates the mean expression level for each gene and each dot represents one gene. Genes with significantly increased expression in the A549 cell line relative to the H460 cell line was indicated. (D) Results of Kyoto Encyclopaedia of Genes and Genomes pathway enrichment analysis.
![]()
The cytotoxic effects of BC-LI-0186 were measured by flow cytometry using annexin V and PI staining. BC-LI-0186 strongly induced H460 apoptosis, whereas A549 cells were BC-LI-0186 resistant (Fig. 2B). To verify that activation of the MAPK pathway confers resistance to BC-LI-0186, DEG analysis was performed in A549 and H460 cells. Overall, 9,197 significant DEGs were identified: 4,001 upregulated and 5,196 downregulated genes (Fig. 2C and D). The upregulated genes in the relatively BC-LI-0186-resistant A549 cells, as compared to H460 cells, were enriched for the following Kyoto Encyclopaedia of Genes and Genomes pathways, in this order: adherens junction organization, cell-cell junction assembly/organization, and MAPK activation. Genes activating the MAPK pathway, such as EGF, FGF14, NTRK1, and RAPGEF2, were among the top DEGs (S2 Table). These activated genes and pathways suggested association of MAPK signaling and BC-LI-0186 resistance, and that targeting MAPK may support cancer treatment.
3. Synergy of trametinib and BC-LI-0186
Therefore, the effect of combining trametinib, which targets MEK1/2, and BC-LI-0186 was evaluated. First, the dose-dependent effects of trametinib on cells were examined. Treatment with more than 1 nM trametinib abolished MEK and ERK phosphorylation (Fig. 3A).
Fig. 3
The combination of BC-LI-0186 and trametinib shows a synergistic effect in human lung cancer cell lines. (A) Immunoblotting of pS6, pMEK, and pERK according to trametinib concentration in the A549 and H460 cell lines. (B) Colocalization images of LC3A/B and LAMP2, and histogram for the ratio of colocalization obtained from slides with 10 or more cells according to the use of BC-LI-0186 and trametinib in A549 and H460 cells. (C) Expression of p62, cleaved poly(ADP-ribose) polymerase (PARP), and cleaved caspase 3 according to the use of BC-LI-0186 and trametinib in A549 and H460 cells. (D) Immunoblotting results pAKT, pS6, pMEK, and pERK according to the use of BC-LI-0186 and trametinib. (E) Fa-dose plot and combination index plot using for treatment of A549, H460, H356, H1703, and H1650 cells.
![]()
Next, we evaluated changes in expression and colocalization of autophagy markers induced by trametinib and BC-LI-0186 using immunofluorescent staining and immunoblotting (Fig. 3B and C). An increased number of LC3A/B puncta and their colocalization with lysosome-associated membrane protein 2 (LAMP2) was induced by BC-LI-0186 in cells cultured in FBS-supplemented media. The combination treatment significantly enhanced these findings. Interestingly, trametinib treatment alone increased the number of LC3A/B puncta and their colocalization with LAMP2 in A549 cells (p=0.0174). These findings were further confirmed under the same culture conditions by immunoblotting for p62, which is associated with mTORC1 and autophagy. Although, both BC-LI-0186 and trametinib increased the expression of p62 in both cell lines, the expression of p62 significantly increased when used in combination. Additionally, there were no differences in the expression of cleaved poly(ADP-ribose) polymerase or cleaved caspase 3, which are markers of apoptosis (Fig. 3C). Finally, the combined effects of BC-LI-0186 (2 μM) and trametinib (10 nM) on A549 and H460 cells were examined (Fig. 3D): MEK, ERK, and S6 phosphorylation decreased in both cell lines, and the paradoxical phosphorylation of S6 induced by BC-LI-0186 treatment alone was absent with the combined treatment.
To clarify whether the two drugs acted synergistically, the CI was obtained through various fixed-dose combinations of BC-LI-0186 and trametinib, using the Chou-Talalay method for drug combination [15]. Fig. 3E displays representative Fa-dose plots and CI plots for BC-LI-0186 and trametinib in A549 and H460 cells. In the combination therapy, the ratio of BC-LI-0186 to trametinib was 1:0.5 in A549 cells and 1:2 in H460 cells. The combination therapy demonstrated strong synergic effect in both cell types. In addition, we examined whether there was a synergic effect in other cancer cell lines, and there was no synergic effect in H1299, H1975, and SNU1330 cell lines, but a synergic effect was confirmed in H358, H1703, and H1650 cell lines. The ratio of BC-LI-0186 to trametinib was 1:0.5 in these cell lines (Fig. 3E).
4. Anticancer effect of the combination of BC-LI-0186 and trametinib using a nude mouse lung cancer xenograft
We generated a mouse xenograft model to confirm the effects of the combination of BC-LI-0186 and trametinib in 9-week-old BALB/c lung cancer model nude mice. Fig. 4A shows a diagram of the treatment schedule. Tumors were measured with callipers and the volume calculated using a modified ellipsoidal formula. TV changes before and after treatment were compared among the groups treated with vehicle, trametinib, BC-LI-0186, or the combination (Fig. 4B). Treatment with trametinib, BC-LI-0186, or their combination significantly reduced TV. The highest increase rate of TV was observed in the vehicle group, while the lowest increase rate was seen in the combination treatment group. A statistically significant difference (p=0.004) was observed between the vehicle group and the combination group, as shown in Fig. 4C. To evaluate the anti-tumor effect of the combination treatment further, tumor tissue was harvested after treatment, and haematoxylin/eosin and immunohistochemical staining for activated caspase-3, pMEK, and pS6 were performed. The number of activated caspase-3 spots was the highest in the combination group (p < 0.001) (Fig. 4D). Compared to the other treatments, BC-LI-0186 had little effect on pMEK expression, while trametinib group had little effect on pS6 expression. However, the combined drugs had a significant effect on both pMEK and pS6 (Fig. 4D). Additionally, there was no significant change in body weight after treatment in any of the groups (Fig. 4E).
Fig. 4
Synergistic anti-tumor effect of BC-LI-0186 and trametinib combination treatment. (A) Diagram of the treatment schedule in BALB/c nude mice lung cancer model. (B) Photographs of typical BALB/c nude mice bearing tumors. (C) Change rate in tumor volume of each group according to differences before and after treatment. There was a significant tumor volume change in control group comparing with combination group (p=0.004). (D) Representative photograph of haematoxylin and eosin and immunohistochemistry staining for activated caspase-3, pMEK, and pS6 on lung sections obtained from mice after 2 weeks’ treatment, and quantification of caspase-3, pMEK, and pS6 in the tumor of each treatment group. *p < 0.05. p-values were obtained by one-way analysis of variance followed by Tukey’s post-hoc multiple comparison tests. (E) Body weight change rate among mouse groups during the course of treatment.
![]()
Discussion
Lung cancer progression through the mTOR or RAS/RAF/MEK pathway is commonly investigated in lung cancer patients without a targetable driver mutation, or those who show no response to targeted therapy or immunotherapy [16,17]. We showed that using a BC-LI-0186/trametinib combination could reduce drug resistance and enhance the therapeutic effect in lung cancer cell line and mouse xenograft experiments.
The constituents of RAS/RAF/MEK and mTOR pathways are frequently activated in various human cancers, leading to sustained tumor growth and treatment resistance [18]. Therefore, when one pathway is targeted by treatment, the other pathway is likely to be mainly responsible for bypassing treatment effects. Simultaneous inhibition of the PI3K/mTOR and RAS/RAF/MEK pathways is currently being investigated for treatment of advanced solid cancers [19–23].
Previous studies have investigated the use of trametinib, a selective allosteric inhibitor of MEK1/2, in lung cancer [21,24]. Blumenshein et al. [24] divided 129 advanced KRAS-mutant NSCLC patients into a trametinib and a docetaxel arm, and evaluated progression-free survival (PFS). Patients in the trametinib arm showed a PFS of 12 weeks (p=0.5197) and similar response rates as those in the docetaxel arm. They suggested that it is necessary to find a KRAS patient subset in whom trametinib will be effective and that a strategy for combining trametinib with other drugs should be developed [24].
Grilley-Olson et al. [21] reported on combining trametinib with GSK2126458 (pan-PI3K/mTOR inhibitor) in 57 advanced solid cancer patients, including lung cancer patients. Although one patient showed partial response and 12 patients showed stable disease for more than 16 weeks, the side effects were unbearable, with rash in 74% and diarrhoea in 61% of patients [21]. The above-mentioned studies suggest the need for combination therapy that can inhibit both MEK/ERK and mTOR, but with fewer side effects. In the current study, when an NSCLC cell line was treated with BC-LI-0186, activation of the MEK and ERK pathways was confirmed. Moreover, cancer was not inhibited by BC-LI-0186 alone, due to the bypass pathway, similar to the results of a previous study [9,25]. However, when we used a combination of BC-LI-0186 and trametinib in a mouse model, lung cancer was effectively suppressed and the side effects were minimal. Fig. 5 illustrates the signal pathway through which BC-LI-0186 and trametinib act, and demonstrates that the combination therapy effectively suppresses lung cancer. The figure shows that BC-LI-0186 and trametinib inhibit the LARS1 and MEK pathway and induce autophagy in lung cancer cells. The combination therapy resulted in a synergistic effect and significantly suppressed the growth of lung cancer cells, as demonstrated by in vivo experiments using mice models.
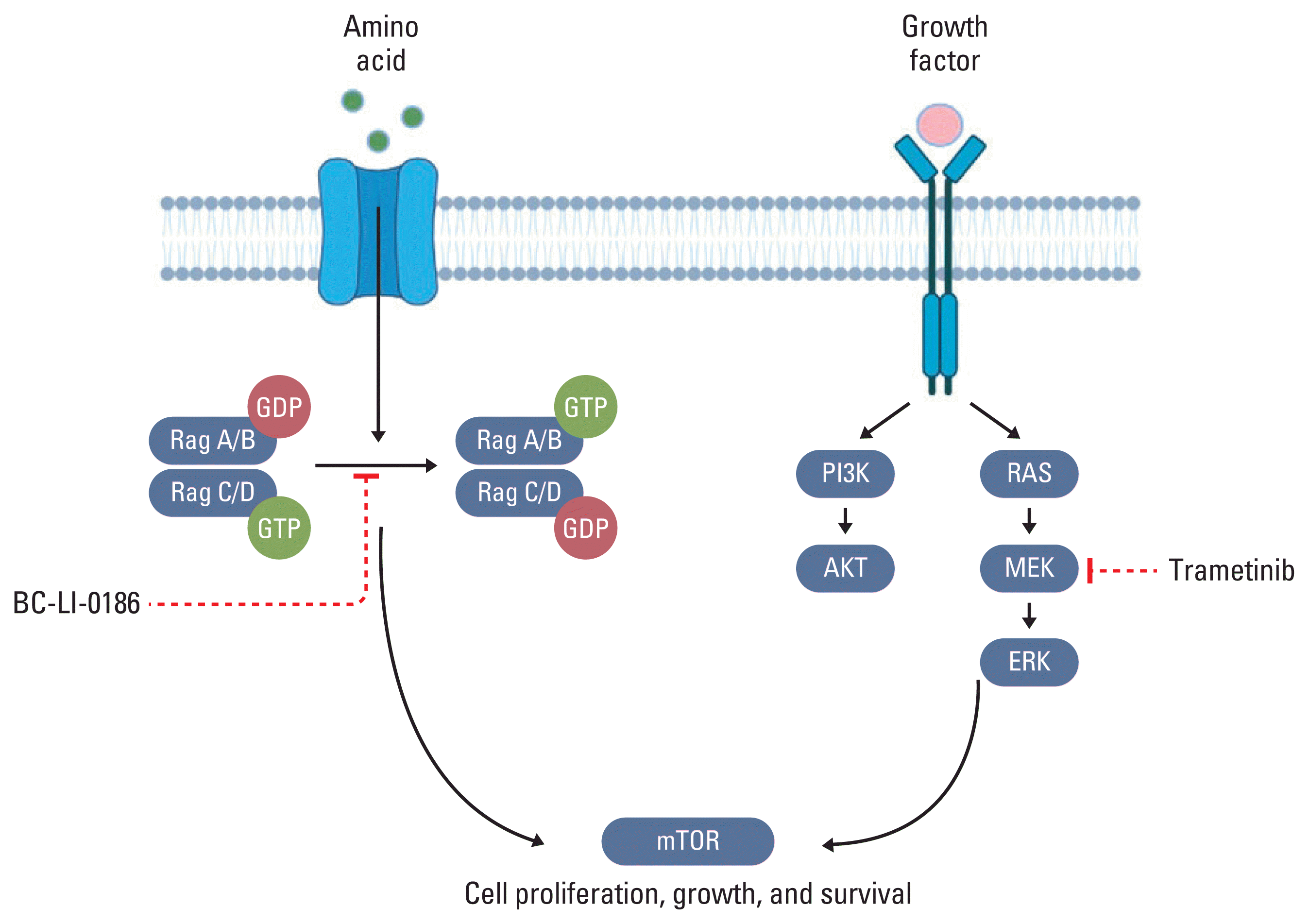
Fig. 5
Schematic diagram of effective cancer suppression process through combination therapy of BC-LI-0186 and trametinib (created in BioRendor.com). mTOR, mammalian target of rapamycin; PI3K, phosphoinositide 3-kinase; Rag, Ras-related GTP-binding protein.
![]()
Because lung cancer is complex and heterogeneous, it is necessary to identify groups that would benefit from the combination treatment [26]. Additionally, TP53 mutations have been shown to confer resistance to BC-LI-0186 in previous studies. Therefore, additional investigations of combined mutations, such as TP53 mutations, are needed to identify the responder group.
In NSCLC cell lines cultured in growth media supplemented with 5% FBS, BC-LI-0186 showed a paradoxical response and activation of the MEK and ERK pathways. When BC-LI-0186 and trametinib were used together, a synergistic anti-tumor effect was observed in a BALB/c nude mouse xenograft cancer model. Thus, the combination of BC-LI-0186 and trametinib inhibits both the mTORC1 and MER/ERK pathways and provides a novel therapeutic strategy for NSCLC.
 XML Download
XML Download