

 PDF
PDF Citation
Citation Print
Print



Introduction
Epidermal growth factor (EGF) and its receptor (EGFR) participate in epithelial cell proliferation and differentiation, thereby promoting wound healing process in normal physiological conditions [1]. Radiotherapy-induced oral mucositis (RIOM) occurs in at least 90% of patients with head and neck cancer (HNC) receiving radiotherapy (RT) as demonstrated by several phase II prospective trials on the therapeutic effect of recombinant human EGF [2–4]. Continuous EGFR signaling due to overexpression or activating mutation causes unregulated cell proliferation and initiates cancer development [5]. EGFR expression has been related to various cancers, including gliomas, HNC, lung, gastrointestinal tract, and urinary system cancers [6]. As approximately 90% of squamous cell carcinoma of HNC and 60% of non–small cell lung cancers overexpress EGFR [7,8], several EGFR inhibitors, such as tyrosine kinase inhibitors or monoclonal antibodies, have been developed as therapies [9]. Unfortunately, EGFR-targeted therapies have limited efficacy and cancers acquire resistance against them [9,10]. Therefore, we need innovative drugs against EGFR-overexpressing cancer cells.
EGF-induced apoptosis in A431 cells, an EGFR-overexpressing cell line, was first reported in 1981 [11]. This phenomenon is also observed in other EGFR-overexpressing and some EGFR-undetectable cell lines [12]. In addition, our team demonstrated a synergistic anticancer activity after EGF administration and irradiation using in vitro and in vivo models [13,14]. If this paradoxical feature of EGF could be manipulated, it could prevent or alleviate RIOM and simultaneously increase tumor cell killing by RT. Currently, the mechanisms underlying EGF-induced apoptosis remain unclear and most of the previous studies are phenomenological observations. The Janus kinase/signal transducer and activator of transcription (JAK/STAT) pathway and mitogen-activated protein kinase kinases/extracellular signal-regulated protein kinase (MEK/ERK) pathway could be involved in EGF-induced apoptosis [15–18]. In this study, we used the clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 (Cas9) knockout screening to understand EGF-induced apoptosis at the genome level. This rapidly evolving technology can edit a specific piece of DNA sequence and allow us to evaluate the effect of the alteration under a biological challenge, such as drug treatment [19].
Based on the CRISPR/Cas9 knockout screening results, we identified dual-specificity phosphatase 1 (DUSP1) as the key molecule in EGF-induced apoptosis. To improve reliability, we assessed DUSP1 mRNA expression in patients with cancer using an open-access dataset in the cBioPortal for Cancer Genomics.
Materials and Methods
1. Cell lines and study drug
EGFR-overexpressing cancer (A431 and A549) and normal control (HEK293FT) cell lines were purchased from American Type Culture Collection (Manassas, VA). The cell lines were cultured in Dulbecco’s modified Eagle’s medium (Gibco BRL, Grand Island, NY) supplemented with 10% fetal bovine serum (Gibco BRL) and 1% penicillin/streptomycin. Cells were grown at 37°C and 5% CO2 in a controlled humidified incubator. We used recombinant human EGF provided by Daewoong Pharmaceutical Company (Seoul, Korea). The EGF powder was reconstituted in distilled water (1 mg/mL) and stored at −70°C.
2. Trypan blue dye exclusion assay
Cells were treated with EGF (0, 0.1, 1, 10, 100, or 1,000 nM) or trametinib (0.2 nM, Selleck Chemicals LLC, Houston, TX). The cells were incubated for 3 or 4 days without changing the medium. After incubation, cells were detached from the culture dishes using trypsin and stained with trypan blue. Viable cells were colorless and counted using a hemocytometer.
3. CRISPR/Cas9 knockout screening in A431 cells
The human genome-scale CRISPR knockout (GeCKO) library v2 was acquired from Professor Yongsub Kim at the University of Ulsan College of Medicine. A431 cells were infected with lentiCas9-Blast (LentiArray Cas9 Lentivirus, Thermo Fisher Scientific, Waltham, MA), and transduction of the Cas9 gene was confirmed through polymerase chain reaction (PCR) (S1A Fig.). We isolated ten monoclonal Cas9-expressing A431 (A431-Cas9) cell populations using limiting dilution. Among them, the sixth monoclonal population was selected for CRISPR/Cas9 knockout screening because its Cas9 intensity was highest on the western blot (S1B Fig.).
Single guide RNAs (sgRNAs) of GeCKO library v2 were packaged into lentiviruses using polyethyleneimine (Sigma-Aldrich, St. Louis, MO) as previously described [20]. After calculating the multiplicity of infection (MOI) for sgRNA lentiviruses (S1C Fig.), A431-Cas9 cells were infected with the pooled sgRNA lentiviruses at an MOI < 1 for 2 days. Post transduction, puromycin selection (8 μg/mL) was performed for 3 days, and the medium was replaced by a normal growth medium without puromycin. After 3 days of recovery, the surviving cells were split into two groups. Each group was treated with 100 nM EGF (experimental group) or an equivalent volume of distilled water (control group), for 3 days. This treatment was repeated four times after each cell had recovered to 70%-90% confluency.
At the end of the final cell recovery period, genomic DNA was harvested using the QIAamp DNA Mini Kit (Qiagen, Valencia, CA) according to the manufacturer’s protocol. Genomic DNA was amplified for next-generation sequencing as described previously [20]. The Illumina HiSeq X Ten platform (Illumina, San Diego, CA) was used for deep sequencing the amplified products. The screen was independently repeated twice. The guide composition of the EGF-treated group was compared with the control group using edgeR. Genes were selected based on the fold change > 2, log2 (normalized data) > 4, and p < 0.05.
4. Short hairpin RNA (shRNA) transfection
For shRNA validation of the top five hit genes, pLKO.1–TRC cloning vector and pLKO.1–TRC control vector were purchased from the Addgene repository (https://www.addgene.org/, plasmid #10878 and #10879). The shRNA oligos were designed using Kay Lab siRNA/shRNA/Oligo Optimal Design (https://web.stanford.edu/group/markkaylab/cgi-bin/), a web-based tool. Each gene was validated with three distinct shRNAs and their sequences are listed in S2 Table. shRNA oligos were cloned into the pLKO.1–TRC cloning vector using the Addgene protocol (https://www.addgene.org/protocols/plko/, accessed on December 3, 2021). Cells were transfected with cloned vectors containing either shRNA or a negative control using the iN-fect in vitro Transfection Reagent (iNtRON Biotechnology, Seongnam, Korea).
5. RNA extraction and reverse transcription–quantitative PCR (RT-qPCR)
Total RNA was extracted using the PureLink RNA Mini Kit (Invitrogen, Carlsbad, CA), reverse transcribed, and amplified using TOPreal SYBR Green RT-qPCR Kit (Enzynomics, Daejeon, Korea) according to the manufacturer’s protocol. A QuantStudio 3 Real-Time PCR instrument (Applied Biosystems, Foster City, CA) was used for qPCR analysis. The primers were synthesized by Macrogen (Seoul, Korea), and their sequences were as follows: DUSP1, forward: CAACCACAAGGCAGACATCAGC, reverse: GTAAGCAAGGCAGATGG-TGGCT; and glyceraldehyde 3-phosphate dehydrogenase (GAPDH), forward: GTCTCCTCTGACTTCAACAGCG, reverse: ACC-ACCCTGTTGCTGTAGCCAA. mRNA expression was calculated using the 2−ΔΔCt method.
6. Western blotting
Western blotting was performed as previously described [13]. Cells were suspended in a cold lysis buffer (iNtRON Biotechnology), sonicated, and centrifuged at 13,000 rpm for 20 minutes at 4°C to obtain cell lysate. The protein concentration of the supernatant was quantified using a bicinchoninic acid protein assay kit (Pierce, Rockford, IL). After protein denaturation using 5× sample buffer (2% sodium dodecyl sulfate, 5% β-mercaptoethanol, 20% glycerol, 0.001% bromophenol-blue, and 0.1 M Tris-HCl [pH 6.8]), protein was separated on 8%-12% Mini-PROTEAN TGX Precast gels (Bio-Rad, Hercules, CA), followed by blotting on polyvinylidene fluoride (PVDF) membranes (Millipore Corp., Billerica, MA). The PVDF membranes were blocked with Tris-buffered saline with Tween 20 (TBST; 10 nM Tris-HCl [pH 7.5], 150 mM NaCl, and 0.1% Tween 20) containing 5% dry milk for 1 hour at room temperature. After washing with TBST, membranes were probed with the following antibodies at 4°C overnight: polyclonal rabbit anti-EGFR (Cell Signaling Technology, Danvers, MA), caspase-3 (Cell Signaling Technology), and β-actin (Santa Cruz Biotechnology, Santa Cruz, CA). Antibodies of ERK (Cell Signaling Technology), p-ERK (Cell Signaling Technology), and DUSP1 (Novus Biologicals, Centennial, CO) were generous gifts from Professor Seung Hee Yang at Seoul National University, Seoul, Korea. These antibodies were also used as the primary antibodies. The dilutions used were 1:2,000 for β-actin antibody and 1:1,000 for others. After incubation with the primary antibody, membranes were incubated for 2 hours at room temperature with peroxidase-conjugated goat anti-rabbit IgG and goat anti-mouse IgG (Jackson ImmunoResearch Laboratories, West Grove, PA) at a 1:1,000 dilution, followed by washing with TBST thrice. Blots were visualized using a WEST-ONE chemiluminescent substrate (iNtRON Biotechnology). Signals were detected using ChemiDoc Touch (Bio-Rad) and quantified using ImageJ (https://imagej.nih.gov/ij/).
7. cBioPortal for Cancer Genomics database
Depending on the EGFR-activating mutations, DUSP1 mRNA expression analysis was performed using cBioPortal for Cancer Genomics (http://www.cbioportal.org/, accessed on 22 September 2022). We included 29 studies with 7,838 samples from the data sets provided by cBioPortal for Cancer Genomics. Of the 29 studies, six dealt with the ‘Head and Neck’ and 23 with ‘Lung’ cancers (S3 Table). After combining the 29 studies, EGFR and DUSP1 genes were queried, and the following parameters were selected for the analysis: mutation type and copy number variation (CNV) of EGFR and mRNA expression (reads per kilobase of transcript per million mapped reads) of DUSP1. Data for 296 samples with DUSP1 mRNA profiles were downloaded. The patients were categorized into two groups: wildtype (WT, n=84) group without mutations and CNV of EGFR, and mutation group (n=39) with known EGFR-activating mutations irrespective of CNV.
8. Statistical analysis
One-way analysis of variance (ANOVA) followed by multiple comparisons test or Student’s t test was used as indicated. All analyses were performed using Prism 8.3.0 (GraphPad Software, San Diego, CA). Illustrations were created using BioRender (https://app.biorender.com/).
Results
1. CRISPR/Cas9 knockout screening for modifiers of EGF-induced apoptosis
EGF-induced cell death was observed in A431 cells in a dose-dependent manner, despite reaching a plateau after 100 nM EGF treatment (Fig. 1A). Therefore, we used 100 nM EGF as the treatment in subsequent experiments. In contrast to A431 cells, the number of viable HEK293FT cells did not decrease after 100 nM EGF treatment (Fig. 1A).
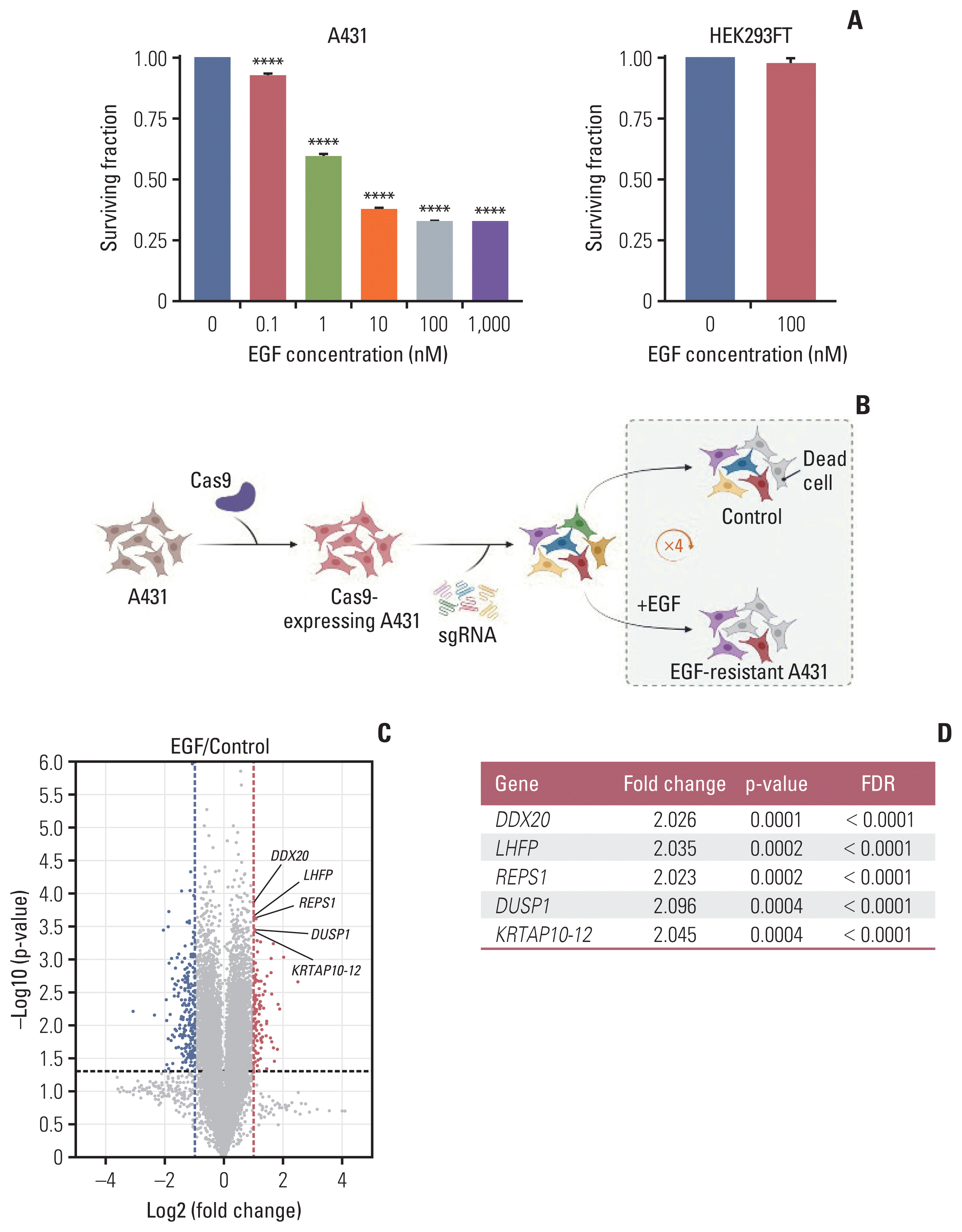
Fig. 1
CRISPR/Cas9 knockout screening using A431 cells. (A) The surviving fraction of cells at varying concentrations of EGF treatment at 3 days (n=3); the EGF treatment range was 0–1,000 nM and 0–100 nM in A431 and HEK293FT cells, respectively. Data are presented as the mean±SEM from one-way ANOVA followed by Dunnett’s multiple comparisons test or Student’s t test, ****p < 0.0001. (B) After sequential transduction of GeCKO library v2 components into A431 cells, A431 cells were split in two groups. The experimental group was treated with EGF (100 nM) and the control group was treated with an equivalent volume of distilled water. To achieve enough EGF-resistant cells, this procedure was repeated four times after every cell confluency recovery to maintain the sgRNA coverage. The screening was performed in duplicates (created with BioRender.com). (C) Volcano plot demonstrating enriched or depleted genes in the EGF screen. Blue dots indicate genes conferring sensitivity and red dots indicate genes conferring resistance to EGF in the gene knockouts. (D) Top five enriched genes according to the p-value and FDR. ANOVA, analysis of variance; Cas9, CRISPR-associated protein 9; CRISPR, clustered regularly interspaced short palindromic repeats; EGF, epidermal growth factor; FDR, false discovery rate; SEM, standard error of mean; sgRNA, single guide RNA.
![]()
We used Human GeCKO library v2 containing 123,411 sgRNAs targeting 19,050 genes (Fig. 1B). To establish A431-Cas9 cells, the A431 cells were transduced with Cas9-expressing lentivirus. We confirmed a decrease in the surviving fraction of A431-Cas9 cells after EGF treatment (S1D Fig.). Subsequently, sgRNAs were transduced into A431-Cas9 cells. To amplify the selection, we added EGF or distilled water to the pooled population of single-gene knockout cells for four pulses. Statistically significant modifiers of EGF-induced apoptosis were detected using the edgeR algorithm. A total of 266 genetic modifiers were identified in gene knockouts: 77 genes were enriched, where sgRNAs protected cells from EGF, and 189 genes were sensitive to EGF and depleted in the pooled population (Fig. 1C).
2. Validation of CRISPR/Cas9 knockout screening
Among the hits from the screening, we selected the top five enriched genes with a false discovery rate cutoff of 5%: DDX20, LHFP, REPS1, DUSP1, and KRTAP10–12 (Fig. 1C and D). To validate the hits, we used an EGFR-overexpressing cell line A549—it is easier to transfect A549 than A431 cells—to demonstrate the versatility of the screening results. A549 cells transfected with shRNA for each of the abovementioned genes were resistant to the cytotoxic effect of EGF compared with A549 cells with negative control shRNA (Fig. 2A). HEK293FT cells were not affected by shRNA transfection and EGF treatment (Fig. 2A).
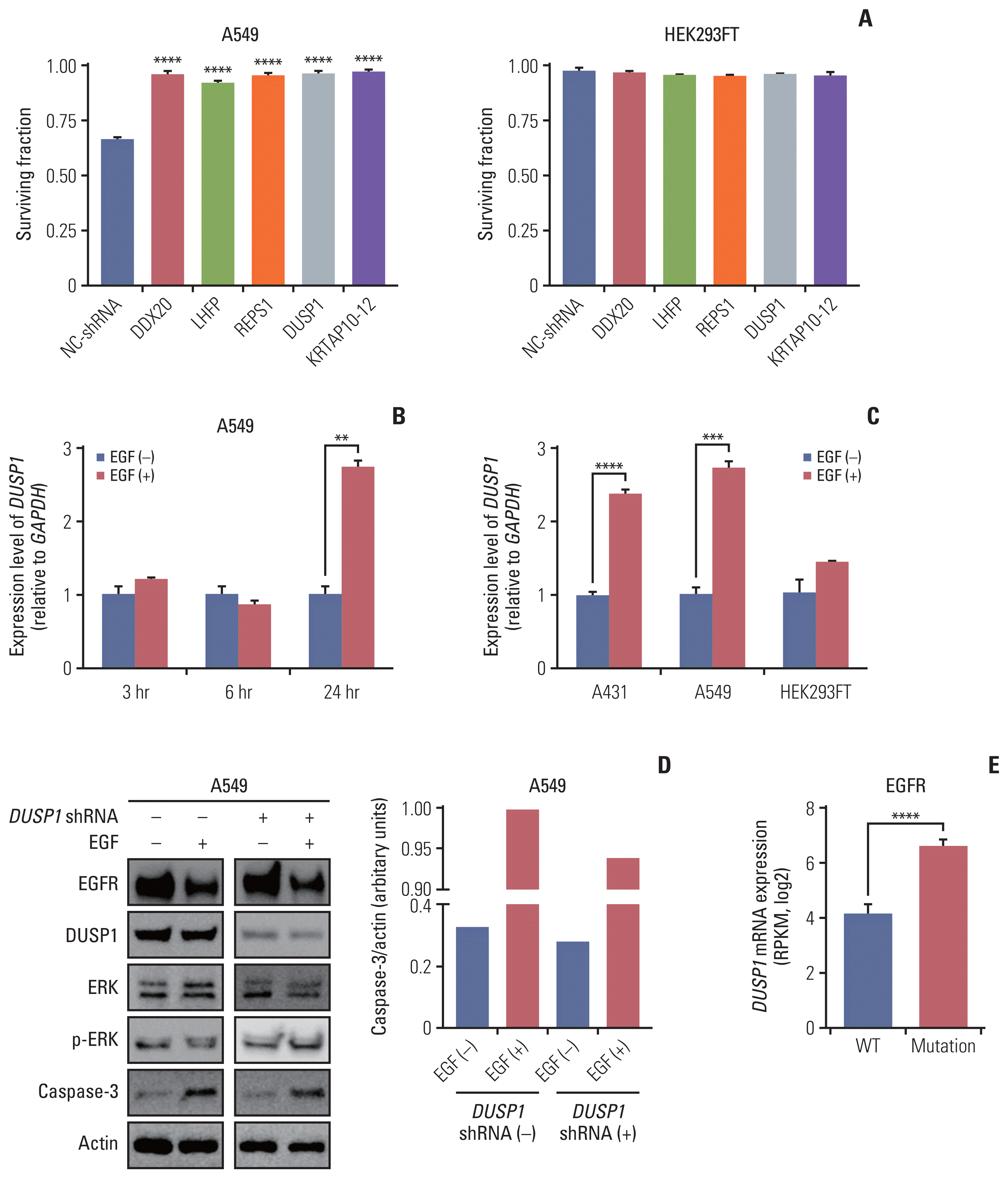
Fig. 2
Validation of CRISPR/Cas9 knockout screening results. (A) Validation of the top five genes using shRNA transfection. The cell surviving fraction was calculated 3 days after EGF treatment (n=3). Each gene was validated with three distinct shRNAs and the graph shows the most efficient shRNA. Data are presented as the mean±SEM from one-way ANOVA followed by Dunnett’s multiple comparisons tests, ****p < 0.0001. (B) The graph showing mRNA levels of DUSP1 quantified using RT-qPCR at three-time intervals: 3, 6, and 24 hours in the control and EGF-treated A459 cells (n=3). (C) The graph showing mRNA levels of DUSP1 quantified using RT-qPCR in the indicated cell lines at 24 h after EGF treatment (n=3). GAPDH was used as a loading control for RT-qPCR. Data are presented as the mean±SEM from Student’s t test, **p < 0.01, ***p < 0.001, ****p < 0.0001. (D) Western blot demonstrating the levels of EGFR, DUSP1, ERK, p-ERK, and caspase-3 in A549 cells. The control and EGF-treated cells were split in two groups: the shRNA-transfected and control (not transfected) groups (n=1). Actin was used as a loading control for western blotting. (E) DUSP1 mRNA levels according to the EGFR-activating mutations in patients with head and neck cancer or lung cancer using the cBioPortal for Cancer Genomics database. Wild type (WT, n=84) indicates both no mutations and no CNV of EGFR, and mutation (n=39) indicates patients with EGFR-activating mutations regardless of the CNV status. Data are presented as the mean±SEM from Student’s t test, ****p < 0.0001. ANOVA, analysis of variance; Cas9, CRISPR-associated protein 9; CNV, copy number variation; CRISPR, clustered regularly interspaced short palindromic repeats; DUSP1, dual-specificity phosphatase 1; EGF, epidermal growth factor; EGFR, EGF receptor; ERK, extracellular signal-regulated protein kinase; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; NC-shRNA, negative control shRNA; RPKM, reads per kilobase of transcript per million mapped reads; RT-qPCR, reverse transcription–quantitative polymerase chain reaction; SEM, standard error of mean; shRNA, short hairpin RNA.
![]()
3. EGF increases DUSP1 mRNA levels in EGFR-overexpressing cell lines
We assumed that one of the above genes was a critical component of EGF-induced apoptosis signaling and involved in apoptosis. Our results for the cell surviving fraction after trametinib treatment, a MEK1/2 inhibitor, with or without EGF (S4 Fig.), proved that this critical component should be associated with upstream or downstream processes of MEK1/2. Moreover, this critical component should serve as a link between the JAK/STAT and MEK/ERK pathways involved in EGF-induced apoptosis [15–18].
After a literature survey, we identified that DUSP1 met all the criteria and other four candidate genes were not associated with JAK/STAT or MEK/ERK pathways. We investigated if EGF induces DUSP1 expression using RT-qPCR (Fig. 2B). In A549 cells, DUSP1 mRNA levels remained unchanged irrespective of EGF until 6 hours after EGF treatment. After 24 hours of EGF treatment, DUSP1 transcription increased significantly in A549 cells, consistent with that observed in A431 cells (Fig. 2C). HEK293FT cells used as controls did not show increased DUSP1 expression after EGF treatment.
4. DUSP1 knockdown reduces apoptosis after EGF treatment
Western blot analysis of DUSP1, phosphorylated ERK, and caspase-3 in WT A549 and DUSP1 shRNA-transfected A549 cells revealed that DUSP1-shRNA significantly reduced DUSP1 protein expression (Fig. 2D). EGF treatment decreased phosphorylated ERK and increased caspase-3 levels in WT cells. In contrast, in DUSP1-depleted cells, EGF treatment increased phosphorylated ERK and reduced caspase-3 levels. Therefore, loss of DUSP1 reduced EGF-induced apoptosis.
5. Patients with EGFR-mutated cancer overexpress DUSP1 mRNA
We analyzed the DUSP1 mRNA levels according to the status of EGFR-activating mutations in 7,838 samples from 29 studies of HNC and lung cancer (downloaded from cBioPortal for Cancer Genomics) to confirm DUSP1 mRNA overexpression due to EGFR gain-of-function mutations. Regardless of the CNV, a substantial increase in DUSP1 mRNA was observed in samples with EGFR-activating mutations compared with the WT (Fig. 2E), confirming EGF-induced DUSP1 expression.
Discussion
EGF has effects on cell growth depending on the cell type. It promotes cell proliferation in normal fibroblasts and inhibits cell growth in EGFR-overexpressing cancer cells [13]. In our study, we reaffirmed this phenomenon in A431, A549, and HEK293FT cell lines. EGFR-overexpressing A431 and A549 cells underwent apoptosis after EGF treatment, even at physiological concentrations of EGF (0.1–1 nM) [21]. However, HEK293FT cells were unaffected by EGF treatment, confirming that EGF-induced cell death is specific to cancer cells. CRISPR/Cas9 knockout screening was used to identify the crucial players involved in this mechanism. We observed that DUSP1 is a promising candidate for a new dual-effect cancer treatment strategy.
In a previous report, EGF-induced apoptosis depended exclusively on the signal transducer and activator of transcription 1 (STAT1) signaling pathway [15]. Another study demonstrated that the EGF-dependent pro-apoptotic response is associated with the p38 mitogen-activated protein kinases (MAPK) present downstream of the EGFR signaling [16]. All these pathways ultimately lead to cell proliferation and survival. However, there existed a missing link connecting these pathways and biasing the signaling to apoptosis. Recently, Ali et al. [18] suggested that EGFR signaling consists of two main pathways: STAT1-dominant (i.e., proapoptosis) and MEK/ERK signaling (i.e., anti-apoptosis). Using trametinib, an allosteric MEK1/2 inhibitor that blocks MEK/ERK, the EGFR signal was transmitted only through the STAT1 pathway, leading to apoptosis with a synergic effect in a metastatic breast cancer cell line, MDA-MB-468 [18]. Based on these results, we conducted a similar experiment using A431, A549, and HEK293FT (S4 Fig.). In contrast to the findings of the study by Ali et al. [18], no synergistic effect of EGF and trametinib was observed, although the statistical significance of this finding could not be analyzed given that only one experiment was performed. The surviving fraction after the combined administration of EGF and trametinib was similar to the lower surviving fraction observed among EGF and trametinib treatment alone. This finding suggests that the missing link should be located in the MEK/ERK pathway.
Among the five hit genes, DUSP1 causes apoptosis by inhibiting p38 MAPK in prostate cancer cells [22]. Gil-Araujo et al. [22] reported this finding and confirmed that apoptosis of DU145 cells increased after DUSP1 overexpression. DUSP1 is a growth factor or stress-inducible gene and suppresses MAPK pathways proteins, such as p38 and ERK, the downstream molecules of MEK [23]. In monocytes and macrophages exposed to interleukin-13, activated STAT1 dimers translocate to the nucleus and enhance DUSP1 expression [24]. Consequently, DUSP1 participates in both the JAK/STAT1 and MEK/ERK pathways. Additionally, activated MAPK activates DUSP transcription as negative feedback, and in the late phase, DUSP enzymatic activity overwhelms that of MAPK [25]. This temporal control of MAPK could explain the relation of MAPK signaling with the apoptosis. Accordingly, we hypothesized that EGFR signaling could lead to DUSP1 expression, further inhibiting ERK and promoting apoptosis. This hypothesis was confirmed using RT-qPCR (Fig. 2B and C) and western blotting (Fig. 2D).
Taken together, we propose the following multistep mechanism to be involved in EGF-induced apoptosis (Fig. 3): (1) EGFR initiates both the JAK/STAT1 and MEK/ERK pathways [15–18]. (2) Phosphorylated STAT1 and negative regulation of ERK prompts DUSP1 expression [24,25]. (3) DUSP1 inhibits ERK activation via MEK [23]. (4) ERK regulation by DUSP1 dominates ERK activation by MEK [25]. (5) Cell signaling is directed towards apoptosis. One possible explanation for this phenomenon in EGFR-overexpressing cancer cells is that as EGFR is abundant in the cell membrane, endocytosis of EGFR will not halt the signals. Because of these persistent signals, DUSP1 activity continues to increase. In patients with EGFR-activating mutation, i.e., incessant EGFR signal propagation, DUSP1 mRNA levels were significantly elevated compared with those in patients with WT EGFR, supporting this explanation.
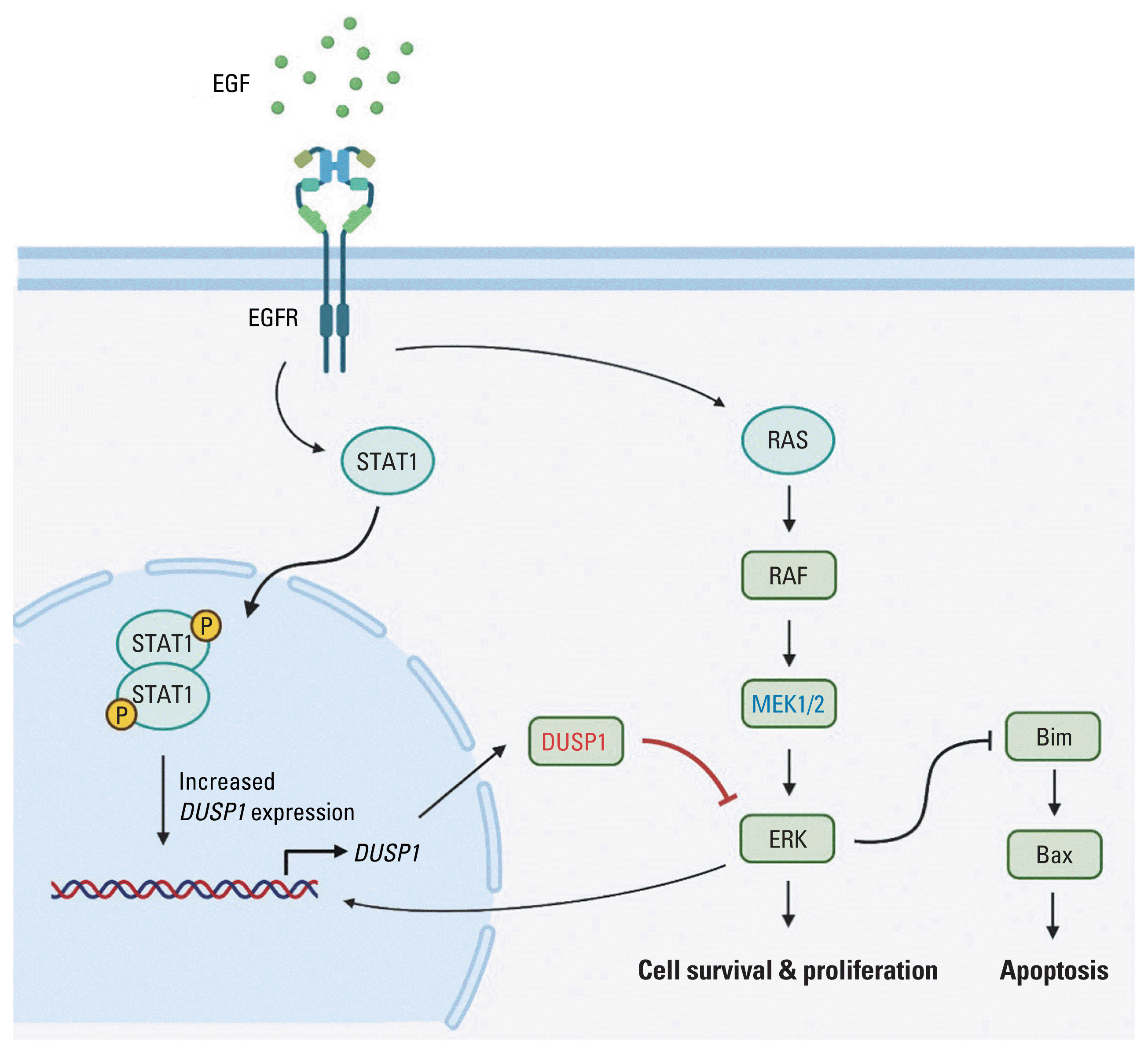
Fig. 3
Proposed mechanism of EGF-induced apoptosis. EGF activates EGFR signaling pathway. EGFR dimerization activates STAT1 dimerization, which then increases DUSP1 expression. DUSP1 inhibits the ERK signaling pathway also activated by EGFR. As a negative feedback, activated ERK increases the expression of its inhibitor, DUSP1. Due to EGFR overexpression and high concentration of EGF, EGFR endocytosis is unable to stop the signaling. Finally, DUSP1 activity overwhelms ERK activity, leading to apoptosis (Created with BioRender.com). DUSP1, dual-specificity phosphatase 1; EGF, epidermal growth factor; EGFR, EGF receptor; ERK, extracellular signal-regulated protein kinase; STAT1, signal transducer and activator of transcription 1.
![]()
Recently, the role of DUSP1 in the biological processes of tumorigenesis has been emphasized. DUSP1 prevents cancer progression in the bladder and hepatocellular carcinoma [26,27]. In contrast, in high-grade serous ovarian cancer, bioinformatics analyses concluded that patients with downregulated DUSP1 had superior progression-free and overall survival rates [28]. To overcome treatment resistance, including that against gefitinib (an EGFR inhibitor), DUSP1 has been considered as a novel therapeutic target [10]. Angiopoietin-like protein 2 exacerbates doxorubicin-induced cardiotoxicity by suppressing DUSP1 [29].
In conclusion, EGF has a paradoxical effect of promoting and inhibiting apoptosis, which may be confined to EGFR-overexpressing cancer cells. We focused on the molecular mechanism involved in EGF-induced apoptosis using an in-depth experimental toolbox, the CRISPR/Cas9 knockout screening system. We discovered 266 genes associated with the sensitivity or resistance to the survival response of EGF. Our data indicated that DUSP1 plays a significant role in EGF-induced apoptosis. DUSP1 should be explored as a promising drug target. Further research is warranted to understand the EGF/EGFR mechanism and epithelial cell-related diseases beyond cancers.
 XML Download
XML Download