

 PDF
PDF Citation
Citation Print
Print



Introduction
Non–small cell lung cancer (NSCLC) accounts for about 80% of all lung cancers globally and has the highest mortality and morbidity rates of any malignant tumor [1,2]. About 40% of patients at presentation have metastatic disease, which is traditionally regarded as incurable. With the development of highly effective systemic therapies, including targeted therapies and immunotherapy, patients with advanced metastatic lung cancer have experienced longer survival times. Regrettably, these systemic treatment options are usually not all curative. In the end, most cancers progress and become fatal, often at sites of known disease [3]. Next to these developments, an emerging area of interest in improving outcome is identification of oligometastatic NSCLC, which has been recognized as a distinct tumor entity that is believed to be early in its evolution of metastatic potential. The term ‘Oligometastasis’ has first been proposed in 1995 by Hellman and Weichselbaum [4], describing a clinically significant state with limited metastatic burden in a single or a limited number of organs. This stage of the disease seems to have an indolent course, better prognosis, and can therefore be considered for radical multimodality treatment. Current definitions of oligometastasis are defined solely numerical as less than three to five metastatic areas in most studies [5], which are imprecise because of ignoring the biological heterogeneity caused by genomic characteristics. With the widespread utilization of more accurate and sensitive staging methods such as fluorodeoxyglucose–positron emission tomography–computed tomography, an increasing number of patients presenting this transitional state between strictly localized and widespread systemic disease have been detected. However, there are still no clinically biomarkers or other techniques to identify oligometastasis that can distinguish the real oligometastatic state from a rapidly progressing disease and thus lead to overtreatment in a subgroup of patients due to the direct initiation of local radical treatment. Identification of patients truly present with an intermediate metastatic state poses clinical diagnostic challenges. Therefore, exploring the genomic features of oligometastatic NSCLC and elucidating its potential difference from the polymetastasis will enable a more thorough selection of patients with a low metastatic burden in whom local radical treatment integrated into multimodality regime can be applied to all metastatic sites.
Systemic therapies, including targeted therapies and immunotherapy, combined with local treatments such as surgery, stereotactic ablative radiotherapy, or thermal ablation can indeed obtain a long-term survival with promising progression-free survival or overall survival [6–9] in oligometastatic NSCLC. Previous research on actionable genomic alterations for targeted therapies and predictive biomarkers of immunotherapy mainly focused on populations of conventional NSCLC. As a result, the frequency and evidence of actionability for all identified somatic alterations and predictor of response to immune checkpoint inhibitors (ICIs) in oligometastatic NSCLC remain unclear. In the precision medicine era, the present best treatment for tumor is individual comprehensive therapy under the guidance of incredible complexity genomics should be realized. Therefore, a reassessment on the landscape and clinical significance of targetable alterations and predictive biomarkers of immunotherapy in patients with oligometastatic NSCLC is important to evaluate the application value of tyrosine kinase inhibitors (TKIs) and ICIs in this group of patients.
In this study, we characterize the genomic features, actionable alterations, predictive biomarkers of immunotherapy in oligometastatic NSCLC by performing 1021-gene next-generation sequencing (NGS), including cancer genetic risk genes, targeted drugs approved by Food and Drug Administration (FDA) or clinical trials, chemotherapy-related genes and prognosis genes. Our results provide considerations for clinical practice and future perspectives that will ultimately improve the selection of patients with real oligometastatic state and optimize personalized cancer therapy for oligometastatic NSCLC.
Materials and Methods
1. Study population and sample collection
We recruited 77 oligometastatic and 21 polymetastatic NSCLC patients from Tumor Center of the Second Affiliated Hospital of Chongqing Medical University between February 2017 and November 2020. Recruited oligometastatic NSCLC patients met the following criteria: (1) had pathologically confirmed NSCLC, (2) had stage IV disease according to the eighth edition of the American Joint Committee on Cancer staging system, (3) had three or fewer metastases within a single organ, not including the primary tumor, and (4) maintain oligometastatic status for three or more months. Eligible polymetastatic NSCLC patients: meet (1), (2) but had more than five metastases in more than three organs. Information on clinical and histopathological characteristics was obtained through electronic medical records. Chongqing Medical University’s ethical committee approved this study, and each participant was required to provide written approval before enrolling.
2. DNA extraction and target capture sequencing
The Geneplus-Beijing Institute (Beijing, China) sequenced tissue samples from all patients using a panel of 1021 cancer-related genes. The tissue source of oligometastasis were all from metastatic lesions, while the tissue source of polymetastasis were all from primary tumors. The online S1 Table lists these 1021 sequenced genes. QIAamp DNA Formalin-Fixed and Paraffin-Embedded (FFPE) Tissue & Blood Mini Kit (Qiagen, Hilden, Germany) was used to extract genomic DNA from FFPE tissue samples and peripheral blood lymphocytes (PBL). DNA concentrations in tissue samples were measured with a Qubit fluorometer and the Qubit dsDNA HS (High Sensitivity) Assay Kit (Invitrogen, Carlsbad, CA), while DNA concentrations in PBL were with the Qubit 3.0 fluorometer and the Qubit dsDNA HS (High Sensitivity) Assay Kit (Thermo Fisher Scientific Inc., Carlsbad, CA). The custom-designed biotinylated oligonucleotide probes (Roche NimbleGen, Madison, WI) covering ~1.4 Mbp coding region of genomic sequence of 1021 cancer-related genes (S1 Table) were designed. A Covaris S2 ultrasonicator (Covaris, Woburn, MA) was used to shear 1.0*g of PBL and tissue DNA into 300-bp fragments for library construction. KAPA DNA Library Preparation Kit (Kapa Biosystems, Wilmington, MA) was used to prepare libraries. An Agilent 2100 Bioanalyzer and an Applied Biosystems 7500 real-time PCR system (Thermo Fisher Scientific Inc.) were used to measure the libraries. DNA was sequenced using an Illumina HiSeq3000 Sequencing System (San Diego, CA) with 2×100 bp paired-end reads. A total of 56 genes were classified as being related to DNA damage repair (DDR), which were grouped into functional pathways, based on literature review and expert curation (S2 Table).
3. Sequencing data analysis
The raw sequencing data were filtered to remove low-quality reads and terminal adaptor sequences. BWA (a Burrows-Wheeler aligner) was used to align the reads to the human genome build GRCh37 [10]. In order to mark PCR duplicates, we used Picard tools (http://broadinstitute.github.io/picard/). MuTect (ver. 1.1.4) [11] and GATK (ver. 3.4–46-gbc02625) [12] were used to call single nucleotide variations (SNVs) and small insertions and deletions (InDels), respectively. Germline variations were filtered using PBL sequence results. The Integrative Genomics Viewer (IGV) [13] was used to manually review all candidate somatic mutations identified by the bioinformatics pipeline by assessing the mapping quality of the reads, the overall read depth at each mutation site, and the quality of the base calls. In order to identify the mutated protein-coding position, ANNOVAR software [14] was used to annotate the gene mutations and filter out intronic and silent changes. A variant allele fraction (VAF) is calculated as follows: sequencing read count of altered alleles/(sequencing read count of reference alleles+sequencing read count of altered alleles)×100%. Mutations in tissue were detected based on the following standards: VAF ≥ 1.0%, and at least five high-quality reads (Phred score ≥ 30, mapping quality ≥ 30, and without paired-end reads bias).
4. Mutational signature analysis
Mutation signatures were defined by analyzing synonymous and non-synonymous somatic SNVs, including six categories of base substitutions, namely, C>A, C>G, C>T, T>A, T>C, T>G, in each included sample. There are 96 substitution types based on the 5′ and 3′ flanking nucleotides of a particular mutant base. Using the 30 signatures documented by Catalogue of Somatic Mutations in Cancer (COSMIC) [15] as references, the potential mutational signatures in each sample were extracted (R package MutationalPatterns) [16]. After that, we analyzed and compared the relative contribution of various signatures in both oligometastatic and polymetastatic tumors.
5. Clinical actionability: OncoKB
Events at the individual gene level were classified based on their therapeutic implications using a precision oncology knowledge database (OncoKB) [17]. This clinical support tool extracts information from published literature to project drug actionability based on existing clinical evidence. It is continuously updated to include emerging biomarker data for FDA-approved protocols, but also those that are still under clinical investigation. A classification system for evidence was established and potentially actionable alterations were classified into one of four levels according to the strength of evidence. Level 1 represents an FDA-recognized biomarker used to predict response to an FDA-approved drug. Level 2 is a standard care biomarker recommended by professional guidelines for predicting response to an FDA-approved drug. Level 3 is compelling clinical evidence supporting the biomarker can predict response to a drug. Level 4 is compelling biological evidence supporting the biomarker can predict response to a drug. “Actionable mutations” were defined as having therapeutic implications, which correspond to OncoKB levels of evidence 1 to 4.
6. Assessment of tumor mutation burden
In tumor tissues, tumor mutation burden (TMB) is defined as the number of somatic mutations and indels per megabyte of coding regions. A TMB score of ≥ 10 mutations/Mb has been proposed as a threshold with a high likelihood of neoantigen formation [18], this was based on the recent FDA approval for pembrolizumab in solid tumors [19]. We divided TMB values into two categories: high TMB (TMB-H), defined as ≥ 10 mutations/Mb; low TMB, defined as < 10 mutations/Mb.
7. Pathogenicity analysis of mutations in DDR genes
DDR genes (including splice site, frameshift, or nonsense) have been classified as deleterious [20]. We used a two-step approach to evaluate the pathogenicity of missense mutations. As a first step, we reviewed all missense mutations identified in the COSMIC and ClinVar [21] databases. As a second step, we used the PolyPhen-2 (Polymorphism Phenotyping v2) prediction tool to evaluate the functional significance of each missense mutation [22]. A missense mutation described as pathogenic by COSMIC and/or ClinVar or with a PolyPhen-2 score of ≥ 0.95 (‘probably damaging’) was considered deleterious. A patient with at least one deleterious DDR mutations is defined as DDR mutant, while a patient with no deleterious DDR mutations is defined as DDR wild.
8. Statistical analysis
ClusterProfiler package [23] was used to explore the biological significance of the somatic mutations of all samples based on the enrichment analysis of gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG). All the statistical analyses were performed using SPSS ver. 26.0 software (IBM Corp., Armonk, NY). The association between any two categorical variables was analyzed using Fisher’s exact test or chi-square test. Any continuous variables were analyzed with the T-test, the Mann-Whitney U-tests or the Kruskal-Wallis rank-sum tests. Trend analysis was represented by p-value, and p < 0.05 indicated statistical significance.
Results
1. Clinical characteristics of patients
Clinical characteristics of 98 lung cancer patients enrolled in this study were summarized in Table 1. Among these patients, 77 were oligometastatic (including 31 bone-only oligometastasis, 28 liver-only oligometastasis, 13 brain-only oligometastasis, and 5 adrenal-only oligometastasis) and 21 were polymetastatic. The median age for all oligometastatic patients was 63 years (range, 33 to 87 years), while for polymetastatic patients the median age was 57 years (range, 46 to 76 years). The median number of metastatic lesions was one in oligometastasis and seven in polymetastasis. Most oligometastatic NSCLC patients were men (41/77), adenocarcinoma (74/77), stage IVa (68/77), smokers (19/45), no family history (43/51), and N2/N3 lymph nodal status (62/77). The echinoderm microtubule-associated protein-like 4 (EML4)–anaplastic lymphoma kinase (ALK) fusions were less (2/77, 2.6%) and epidermal growth factor receptor (EGFR) mutation (43/77, 55.8%) were more commonly found. The majority of patients with polymetastatic NSCLC were female (10/21), non-smokers (7/14), no family history (14/15) and N2/N3 lymph node status (19/21), while all polymetastasis presented with stage IVb. The EGFR mutation and EML4-ALK fusions were 61.9% (13/21) and 9.5% (2/21), respectively.
Table 1
Clinical characteristics 77 oligometastatic and 21 polymetastatic NSCLC patients
![]()
2. The discrepancy and concordance of the mutational landscape between oligometastatic and polymetastatic NSCLC
The mutational landscape of somatic alterations in 77 oligometastatic and 21 polymetastatic NSCLC generated from targeted sequencing data is shown in Fig. 1A and B. The most frequently mutated genes (> 10%) in oligometastatic NSCLC were TP53 (64%), followed by EGFR (56%), cyclin dependent kinase inhibitor 2A (CDKN2A; 22%), RB transcriptional corepressor 1 (RB1; 13%), and SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily A, member 4 (SMARCA4; 12%). Except for the most prevalent tumor suppressor and driver genes (TP53 and EGFR), which were relatively consistent between two groups, their high-frequency mutation spectrum differed considerably. As summarized in S3 Table, among the oligometastatic patients, 608 genomic alterations were identified in 232 genes. The most common mutational type was missense (59.54%), followed by amplification (14.14%) and nonsense (7.89%). We also detected 186 genomic alterations in 119 genes among polymetastatic group. Remarkably, oligometastatic tumors had more copy number deletion mutations as compared to polymetastatic tumors (2.96% vs. 0%, p=0.011). We observed only 25.4% genetic mutations were shared, suggesting a high intertumor mutational heterogeneity between oligometastatic and polymetastatic tumors (S4A Fig.). As a result of examining the SNV spectrum, the Ti/Tv ratios of oligometastatic and polymetastatic tumors were 0.67 and 0.88 respectively, with the most frequent base substitution bias toward cytosine (C)>thymine (T) transitions followed by cytosine (C)>adenine (A) transversions in two cohorts (Fig. 1C–E), both of which are associated with exposure to cigarette smoking [24].

Fig. 1
Alteration landscape of 77 oligometastatic (A) and 21 polymetastatic (B) non–small cell lung cancer (NSCLC) patients. The heat map shows top 20 genes across all samples, with genes ranked by mutation frequency. Top bar summarizes the total number of mutations in each patient (columns). Side bar (rows) summarizes the percentage of tumors with mutation in each gene and mutation composition for each gene in the entire cohort. Bottom heat map, group, smoking, sex, and age. Different colors denote different types of mutations and different clinical features. Single nucleotide variations (C), Ti/Tv ratios in oligometastasis (D) and polymetastasis (E), respectively. Differentially mutated genes between oligometastatic and polymetastatic NSCLC (F). The mutual exclusivity and co-occurrence analysis in each group (G, H).
![]()
To identify genes associated with the presence of indolent state, we compared the frequencies of genomic alterations in oligometastatic and polymetastatic cohorts (S5 Table). We observed that Erb-B2 receptor tyrosine kinase 2 (ERBB2; 3.90% vs. 23.81%, p=0.011), ALK (6.49% vs. 23.81%, p=0.035), myeloid/lymphoid or mixed-lineage leukemia 4 (MLL4; 0.00% vs. 9.52%, p=0.044), phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit beta (PIK3CB; 0.00% vs. 9.52%, p=0.044), and DNA topoisomerase II alpha (TOP2A; 0.00% vs. 9.52%, p=0.044) were mutated at a significantly lower frequency in oligometastatic patients (Fig. 1F). We also analyzed the prevalence of copy number variation (CNV) alterations and identified 104 and 23 clinically related CNV events in oligometastasis and polymetastasis, respectively. The most frequently CNV changes in oligometastasis were EGFR amplification (23.38%), following by CDKN2A deletion (14.29%), MYC amplification (9.09%), and cyclin dependent kinase inhibitor 2B (CDKN2B) deletion (9.09%). However, the most common CNV gain in polymetastasis were EGFR (14.29%), ERBB2 (14.29%), forkhead box A1 (FOXA1; 9.52%), and fibroblast growth factor receptor 1 (FGFR1; 9.52%) (S4C and S4D Fig.). Meanwhile, we also searched for genes with a mutation frequency of at least 5% in oligometastasis but extremely low (0%) in polymetastasis and found CDKN2B, phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic sub-unit alpha (PIK3CA), ABL proto-oncogene 1, non-receptor tyrosine kinase (ABL1), BRCA1 DNA repair associated (BRCA1), CREB binding protein (CREBBP), catenin alpha 1 (CTNNA1), and phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit gamma (PIK3CG) as mutated in approximately 10%, 9%, 6%, 5%, 5%, 5%, and 5% among oligometastasis (S5 Table). These low prevalence unique mutations may also be involved in constituting important features of oligometastasis. In addition to the comparison of mutation frequencies, we also analyzed somatic alteration interactions. Co-occurrence and mutual exclusivity analysis revealed that CDKN2A/CDKN2B, CDKN2A/MYC, CDKN2B/MYC, PIK3CA/ RB1, and SMARCA4/LRP1B were significantly co-occurring, but EGFR displayed exclusivity with kelch like ECH associated protein 1 (KEAP1) in oligometastatic tumors (Fig. 1G). However, we only detected EGFR and BRCA1 associated RING domain 1 (BARD1) mutations that were mutually exclusive in polymetastatic tumors (Fig. 1H).
Inconsistent efficacy to targeted therapies has been obser-ved in NSCLC patients with different mutation subtypes and treatment strategies remain to be optimized based on precisely subtype analysis. We further individually analyzed the subtypes of driver gene mutations in oligometastatic NSCLC (S6 Fig.). EGFR p.L858R and exon 19del comprised 21.70% and 16.87% of 83 EGFR mutations, respectively. KRAS p.G12V, p.G12V, and p.G12C made up 50.00%, 25.00%, 25.00% of eight KRAS mutations. The PIK3CA E545K mutation associated with EGFR-TKI resistance accounted for 14.29% of the seven PIK3CA mutations. The most common mutant subtype of MET was amplification (4/7, 57.14%). Fusion driver genes including EML4-ALK fusions and KIF5B-RET fusions were detected in two and one tumor respectively, but we didn’t find ROS1 fusion in any patient. ERBB2 amplification and BRAF V600E mutation were found in one and one tumor, respectively. Considering both groups have a large proportion of EGFR-positive patients, we further compare the occurrence of EGFR 19del and L858R which are canonical targets of EGFR inhibitors. Oligometastatic patients tended to have a lower incidence of EGFR 19del, with borderline significance (18.18% vs. 38.10%, p=0.054), while the missense mutations, EGFR L858R, had a comparable incidence between two series (23.38% vs. 19.05%, p=0.754) (S4B Fig.).
3. Enrichment of somatic mutations by GO and KEGG analysis
To further determine whether these genomic alteration landscapes can lead to difference in cancer-related signaling pathways and biological processes, we performed pathway-level analysis using the KEGG and GO database. For each of the two groups, the genes affected by somatic mutations and/or CNVs were concatenated and tested the potential enrichment against each KEGG pathway. Based on gene count and p-value, Fig. 2 showed the top 15 pathways in each group enriched by KEGG (Fig. 2A) and GO (Fig. 2B). Altered signaling pathways included central carbon metabolism in cancer, phosphoinositide 3-kinase (PI3K)–Akt signaling, FoxO signaling pathway, Rap1 signaling pathway, Ras signaling pathway, p53 signaling, ErbB signaling, vascular endothelial growth factor signaling, and other well-known pathways. We observed both oligometastatic and polymetastatic tumors had strikingly significant enrichment of pathways associated with cell proliferation, invasion, and metastasis, including the PI3K-Akt signaling pathway, Rap1 signaling pathway, and mammalian target of rapamycin signaling pathway, which may be related to the fact that they were all advanced metastatic tumors. Remarkably, compared with polymetastasis, oligometastasis has a unique significant enrichment of apoptosis signaling pathway. Deeply analyze the proportion of specific signaling pathways enriched in patients, oligometastatic patients were also observed significantly harbored hypoxia inducible factor 1 (HIF-1) signaling pathway (64.94% vs. 90.48%, p=0.027) (S7 Table) related to invasion/metastasis than polymetastasis. Consistent with previous results, the differentially mutated genes identified in two groups, such as ERBB2 and PIK3CB, which were substantially more frequent in polymetastasis, participate in HIF-1 pathway. Top GO terms showed that the mutated genes of two groups might be involved in the following common functional categories: peptidyl-tyrosine phosphorylation, protein autophosphorylation, transmembrane receptor protein tyrosine kinase signaling pathway, negative/positive regulation of transcription from RNA polymerase II promoter, cell proliferation, etc. Intriguingly, the cellular behaviors-related biological processes, negative regulation of cell proliferation, was robustly activated in oligometastatic NSCLC.

Fig. 2
Top 15 enriched pathways by Kyoto Encyclopedia of Genes and Genomes (KEGG) (A) and functional terms by GO enrichment (B) of somatic mutations in oligometastatic and polymetastatic non–small cell lung cancer (NSCLC) patients. Count: the number of mutations enriched in this signaling pathway or functional term. GO, gene ontology; HIF-1, hypoxia inducible factor 1; KEGG, Kyoto Encyclopedia of Genes and Genomes; MAPK, mitogen-activated protein kinase; mTOR, mammalian target of rapamycin; NSCLC, non–small cell lung cancer; PI3K, phosphoinositide 3-kinase; VEGF, vascular endothelial growth factor.
![]()
4. Mutational signature analysis
Mutational signatures are reflections of the mutational processes that have been active throughout someone’s life [25]. The mutational signature analysis was performed on all cases using the non-negative matrix factorization method implemented in MutationalPatterns R package [16] to infer the underlying mutational processes. Comparing the mutational patterns between oligometastatic and polymetastatic NSCLC patients, the cosine similarity was only 0.771 which suggested there were potential differences between two groups (Fig. 3A). A total of several mutational signatures (corresponding to COSMIC signatures) were extracted from all samples. These signatures were further compared with the previously defined COSMIC signatures (https://cancer.sanger.ac.uk/cosmic/signatures_v2) in order to determine the clinical relevance of these signatures. Based on the trinucleotide context of the substitutions, the resulting signature was decomposed into 30 signatures for mutational processes in humans, which allows us to evaluate the relative proportion of the mutational processes contributing to the etiology of oligometastatic and polymetastatic NSCLC. We found that approximately 16.61%, 16.50%, 32.40%, 6.48%, 14.19%, 3.94%, and 9.87% of the mutational signatures of oligometastatic NSCLC consisted of COSMIC signature 1 (age-related), signature 3 (failure of DNA double-strand break-repair by homologous recombination), signature 4 (exposure to tobacco mutagens), and signature 6 (DNA mismatch repair defect-related), signature 11 (alkylating agent-related), signature 14 (unknown etiology), and signature 24 (exposure to aflatoxin), respectively, while 18.92%, 43.86%, 11.28%, 11.49%, 9.64%, and 4.80% of the mutational signatures of polymetastatic NSCLC consisted of COSMIC signature 1, signature 3, signature 4 and signature 6, signature 11 and signature 24, respectively (Fig. 3B). This indicated that the higher frequency of tobacco mutagens exposure appeared to play a strong role in oligometastatic cancers. Meanwhile, the significantly reduced capacity of DNA double-strand break-repair may indispensably participate in promoting extensive tumor metastasis. Additionally, COSMIC signature 14 was only observed in the oligometastatic cohort whose etiology remains unknown.
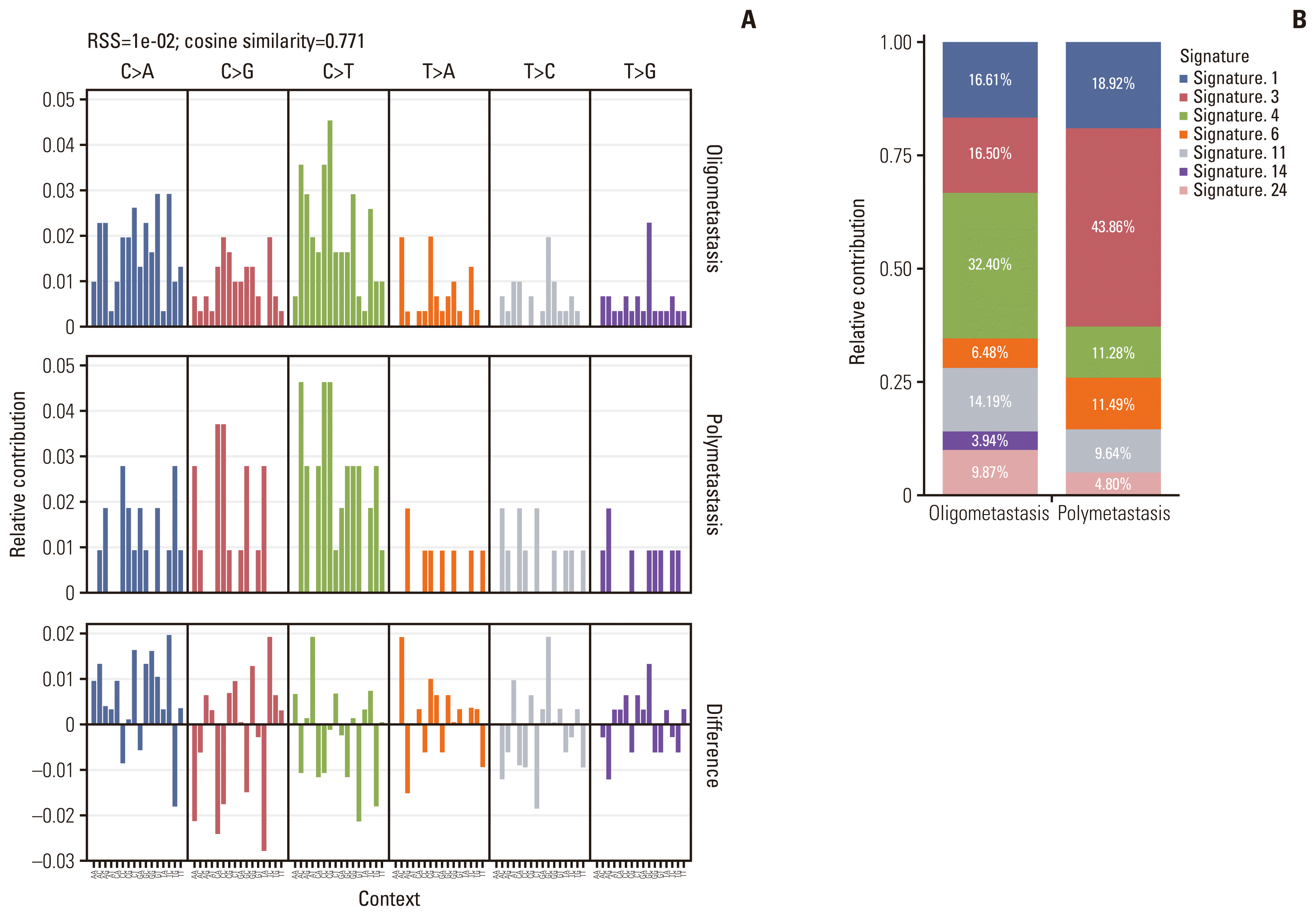
Fig. 3
Mutational signatures in oligometastatic and polymetastatic non–small cell lung cancer (NSCLC) patients. (A) Ninety-six subtype classification of the mutational signature of oligometastatic and polymetastatic NSCLC. (B) Histogram of mutation signatures for oligometastatic and polymetastatic NSCLC patients. RSS, residual sum of squares.
![]()
5. Clinical actionability for targeted therapy
With the development of precision medicine, a variety of new drugs and new progress in targeted therapy of lung cancer emerge in an endless stream, bringing more choices for clinical diagnosis and treatment. Considering that not all somatic changes can result in targetable mutations, we used OncoKB (http://oncokb.org/) to classify mutations of oligometastatic NSCLC patients into different levels based on evidence of clinical actionability. Evidence level was defined as the highest level among all actionable mutations in each patient. Overall, 74.03% of patients had at least one actionable mutation, and three types of evidence levels, including level 1, level 2, and level 4, were identified (Fig. 4A). According to the annotation results, 57.14% tumors harbored level 1 gene alterations including SNVs of KRAS, EGFR, BRAF, and ALK, in-frame shift of EGFR and fusions of RET and ALK. Three point nine percentage tumors harbored level 2 gene alterations including amplifications of MET and ERBB2, fusion of RET, and in-frame shift of ERBB2. Level 4 accounted for 12.99%, including SNVs in PTEN, KRAS, CDKN2A, and BRAF, frameshift of CDKN2A and deletion of CDKN2A (Fig. 4A–C). Of note, the most prevalent genetic changes across all targetable somatic changes were EGFR, followed were CDKN2A and KRAS mutations, and the distribution of actionable mutation spectrum was similar to that in NSCLC cohort in some other studies [26,27]. In our oligometastatic cohort, all the potential drugs targeting specific genetic alterations were also presented in Fig. 4C.
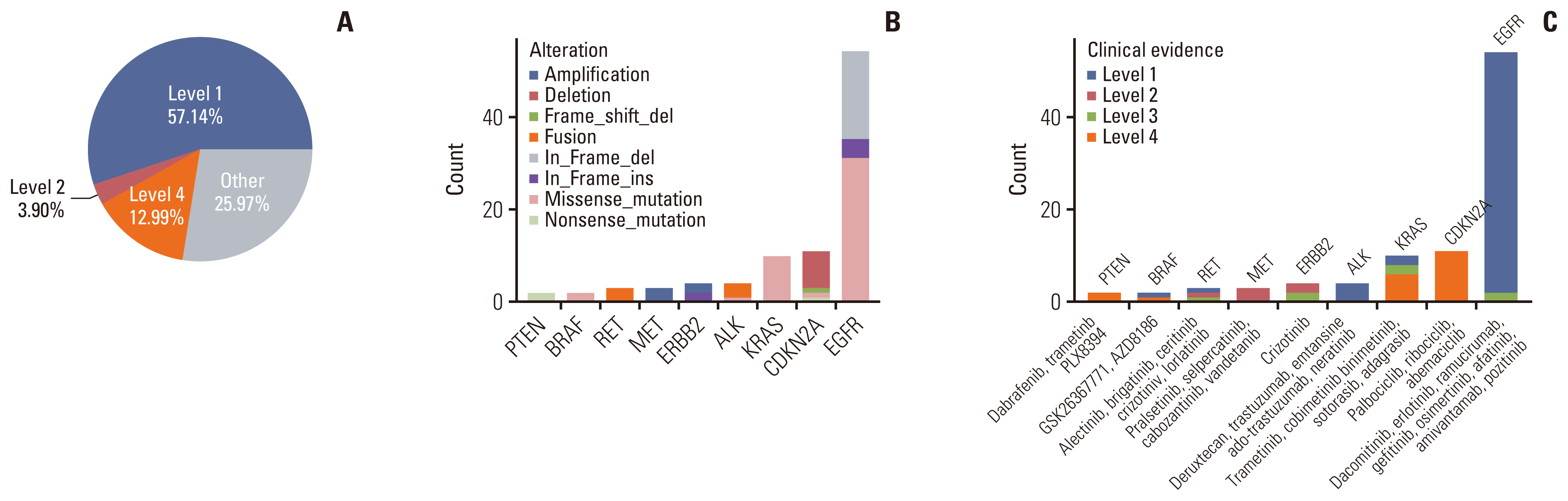
Fig. 4
Somatic alterations identified by the 1021-panel that are clinically actionable in oligometastatic group. (A) Samples were classified according to their highest level of actionable alterations. (B) Distribution of alteration types. (C) Distribution of levels of actionable alterations. ALK, anaplastic lymphoma kinase; BRAF, B-Raf proto-oncogene, serine/threonine kinase; CDKN2A, cyclin dependent kinase inhibitor 2A; EGFR, epidermal growth factor receptor; ERBB2, Erb-B2 receptor tyrosine kinase 2; PTEN, phosphatase and tensin homolog.
![]()
6. TMB and its clinical correlation
In recent years, the treatment and prognosis of NSCLC has rapidly benefited from ICI treatment. TMB-H is emerging as an independent predictor of response to ICIs, such as programmed cell death 1 and programmed death-ligand 1 [28]. In general, low TMB renders response to immunotherapy less likely [29]. Previous studies have demonstrated that NGS panels can be a reliable technology to analyze TMB [30,31]. Based on measurements of available TMB in 42 oligometastatic and nine polymetastatic cases, the median TMB in oligometastatic series was 5.00 mutations/Mb (range, 0.00 to 72.00 mutations/Mb) and 6.00 mutations/Mb (range, 2.88 to 19.00 mutations/Mb) for polymetastatic series, with no significant difference (Fig. 5B). Further analyzed the oligometastatic NSCLC patients, the 14.29% of cases (6/42) who had TMB-H and may benefit from ICI treatment were shown in Fig. 5A. Historically, it has been reported that there was a strong positive correlation between DDR gene mutations and responses to immunotherapy in NSCLC. A defect in DNA replication can increase the rate of somatic mutations and lead to a higher TMB [31]. As expected, oligometastatic lung cancers with somatic mutations in DDR genes got higher TMB than wild-type cancers (median value, 5.76 vs. 4.00 mutations/Mb; p=0.022) (Fig. 5D). Interestingly, we identified one patient, a current smoker, which showed a high TMB of 72 mutations/Mb, significantly deviating from a normal distribution (Fig. 5A). Many known DDR genes, such as ATR, BARD1, BRCA1, CDK12, CUL3, FANCL, MUTYH, TP53, and WRN, were among the mutated genes in this sample. Furthermore, the median TMB was higher in smokers than non-smokers in oligometastasis (median value, 20.16 vs. 4.00 mutations/Mb; p=0.003) (Fig. 5C). However, there was no significant difference between TMB and age, sex and metastatic site (S8A–C Fig.).
Fig. 5
(A) Distribution of tumor mutation burden (TMB) in 42 oligometastatic and nine polymetastatic non–small cell lung cancer patients. Non-parametric test of TMB according to metastatic state (B), smoking status (C), DNA damage repair (DDR) genes (D), TP53 genotype (E), SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily A, member 4 (SMARCA4) genotype (F), LDL receptor related protein 1B (LRP1B) genotype (G), and ABL1 genotype (H).
![]()
We next investigated the association of high-frequency mutated genes with TMB. Patients were divided into wild-type and mutant groups according to the mutation status for specific genes. Particularly, the most common mutation in oligometastatic patients is TP53, which may be a useful biomarker for predicting the response to immunotherapy in different cancer types [32]. The median TMB for TP53-mutant tumors was 5.76 mutations/Mb, which was significantly higher than that for wild-type TP53 tumors (4.00 mutations/Mb, p=0.033) (Fig. 5E). Previous studies have shown that CDKN2A mutation is significantly correlated with TMB in lung cancer [33], while EGFR mutation is significantly correlated with low TMB [34]. Considering these two mutations are both critical top genetic variations of oligometastasis, we further analyzed the relationship between these two genes and TMB. However, our cohort showed that mutations in EGFR and CDKN2A were not significantly associated with TMB in oligometastatic NSCLC patients (S8D and S8E Fig.). Also of note, LRP1B and SMARCA4, two other high-frequency mutant genes in oligometastatic group, were reported to be associated with higher TMB and survival outcome in NSCLC in the previous study [35,36]. LRP1B-mutant and SMARCA4-mutant tumors had higher TMB than those with wild type (median value, 20.16 vs. 4.80 mutations/Mb; p=0.002 and 20.16 vs. 5.00 mutations/Mb, p=0.018) (Fig. 5F and G). In addition, tumors with ABL1 mutations had higher TMB levels vs those with wild type (median value, 20.16 vs. 5.00 mutations/Mb; p=0.049) (Fig. 5H), this was not the case for tumors with RB1, CDKN2B, KRAS, PIK3CA, MET, MYC, and KEAP1 mutations (S8F–L Fig.).
Discussion
In the European Society of Medical Oncology and the National Comprehensive Cancer Network guidelines [37,38], oligometastatic NSCLC was recognized as a special treatment entity. Though the biology and behavior of ‘intermediate state’ of metastatic disease are not well understood, numerous evidences suggested that local radical treatment can benefit the subgroup of patients when integrated into a multimodal treatment regimen. However, until recently, few reports explored genomic characteristics of oligometastatic disease and there are no biomarkers or biology processes that can reliably identify oligometastatic patients with truly limited metastatic capacity. Identification of the real oligometastatic tumors has become a new challenge to clinical diagnosis in the era of precision medicine. Therefore, we investigated genomic profiles of oligometastatic NSCLC and explored its differences from polymetastatic NSCLC in somatic mutations, pathway-level analysis, mutational signature, therapeutic targets, and tumor mutational burdens by NGS to facilitate precise diagnosis and therapy. Remarkably, we found oligometastatic tumors had more copy number deletion mutations than polymetastatic tumors (p=0.011). Since CNV is an important manifestation of chromosomal instability (CIN) and CIN has been reported to be associated with metastasis [39], the important role of CIN in the formation and process of oligometastasis needs to be rigorously investigated. Moreover, we also observed significantly lower frequency of mutations in ERBB2, ALK, MLL4, PIK3CB, and TOP2A in oligometastatic patients, suggesting patients with advanced NSCLC who carry these differentially mutated genes are more likely to be polymetastasis than oligometastasis. Specifically, mutations in MLL4 have been reported to promote both the initiation and progression of cancer and PIK3CB alteration was correlated with increased cancer cell proliferation and promotion tumor growth [40,41]. We also found oligometastatic patients harbored several somatic mutations of certain high-frequency genes including TP53 (64%), EGFR (56%), CDKN2A (22%), RB1 (13%), KRAS (10%), PIK3CA (9%), MET (9%), and NF1 (5%) associated with p53, RTK/RAS, PI3K-Akt and cell cycle pathways, which were reminiscent of the genomic alterations reported in conventional NSCLC [42,43]. Given the important role of these signaling pathways in the feature of cancer proliferation, cell motility, invasion, and cancer metastasis, these variants might be relevant to the weak metastatic ability of oligometastatic NSCLC. Mutual exclusivity and co-occurrence analysis showed that EGFR and KEAP1 alterations were mutually exclusive in oligometastatic NSCLC, suggesting biologically distinct subgroups may frequently be distinguished by these genes. Clinical studies have shown that KEAP1 mutations in NSCLC are associated with resistance to multiple cancer therapies, including chemotherapy, TKI therapy, X-ray irradiation, and shorter overall survival [44,45]. Therefore, the mutually exclusive pattern between EGFR and KEAP1 mutations may accurately stratify oligometastatic NSCLC with different prognoses and response to antitumor therapy.
In our study, the collective somatic mutational signature was identified based on both the type of substitutions and the context of the substitution. Each collective signature of NSCLC from oligometastatic or polymetastatic patients underlies a mutagenic process. Apart from environmental factors such as alkylating agents, tobacco smoking, and exposure to aflatoxin, intrinsic sources, such as DNA double-strand break-repair and DNA repair defects, have been described as the main cause of oligometastatic NSCLC. Moreover, since the proportion of signature 3 and 6 is lower than that of polymetastasis, oligometastatic NSCLC may have a stronger ability to repair DNA damage. Notably, we found that the signature 14 whose etiology remains unknown contributes 3.94% to oligometastasis signature, but it was not observed in polymetastatic cohort, suggesting that there exists a distinctive mutagenic process in oligometastatic NSCLC. Future studies linking this signature of unknown origin with cancer risk factors may provide insights into mechanisms and offer avenues for further research into oligometastatic disease.
As oligometastasis had heterogenous level of SNVs and CNVs in comparison to polymetastatic cohort, we further analyzed their involvement in cancer-related signaling path-ways. In particular, we focused on PI3K-Akt signaling pathway, which plays an important role in carcinogenesis. Overall, 85.90% of oligometastatic NSCLC patients (67/78) had mutations in the PI3K pathway. This is certainly consistent with the situation that multiple PI3K pathway modifier is under clinical study and may be a treatment option for these patients [46–48]. In contrast to polymetastasis, oligometastasis appeared to have an indolent course characterized by “intermediate state of metastasis.” However, the mechanism of maintaining tumor dormancy in oligometastatic NSCLC has not been well characterized. In the current study, we found oligometastatic patients harbored significantly lower frequencies of mutations in genes such as ERBB2 or PIK3CB, which participates in HIF-1 signaling pathway, compared with polymetastatic samples. Downregulation of HIF-1 signaling may be associated with inhibition of distant metastasis, proliferation, and invasion of tumors [49]. Moreover, compared with polymetastatic group, oligometastatic disease has a unique significant enrichment of apoptosis signaling pathway. DNA damage checkpoint gene, ATM, which induce cell death or senescence in response to DNA damage or DNA replication stress [50] and is involved in apoptosis signaling. Mutations in ATM occurred less frequently in oligometastatic patients in comparison with polymetastatic patients (4% vs. 10%, p=0.291). The lower frequency of ATM mutations may promote apoptosis to maintain the indolent state due to a stronger ability to repair DNA damage in oligometastasis. Together, these results possibly explain the relatively weaker state of tumor metastatic potential and possible dormancy in oligometastatic NSCLC compared with polymetastasis. Further studies are necessary to detail such mechanisms and identify targets in these pathways that may control the formation of oligometastasis in NSCLC.
Nowadays, patients with specific molecular changes have greatly benefited from targeted therapy. Treatment decisions should be based on molecular changes in a tumor, with standardized and easily interpreted annotations. OncoKB provides detailed, evidence-based information about individual somatic mutations. In this study, we found 74.03% of patients with oligometastasis had at least one actionable mutation. This percentage was comparable with the 67% reported in 1,564 patients with usual advanced NSCLC in the previous study [51]. However, there were 49.35% of oligometastatic patients carrying EGFR T790M mutations (15.58%) or sensitizing EGFR mutations including 19del (18.18%), L858R (23.38%), L861Q (1.30%), and S768I (1.30%), which were also higher than the conventional advanced NSCLC patients (41.2%) as previously reported [51]. Given the higher frequency of EGFR-sensitive driver gene mutations, this may partly explain a better prognosis of patients with oligometastatic NSCLC. Besides EGFR mutations, the other most common actionable mutations were CDKN2A and KRAS mutations, which were the major components of level 3 and 4 alterations. A level 3 or 4 alteration indicates that there is compelling clinical or biological evidence to support the biomarker as a predictor of response to an existing drug. Given the high frequency and clinical evidence classification of CDKN2A and KRAS actionable alterations in oligometastatic NSCLC, therapeutic developments in CDKN2A or KRAS inhibition would be of great interest. Actually, KRAS mutations have traditionally been considered untargetable until the recent development of KRAS G12C inhibitors, which have shown promise in the treatment of lung cancers and other solid tumors that harbor a KRAS G12C mutation [52,53]. As oncology research and drug development advance rapidly, more oligometastatic patients with level 3 or 4 alterations as well as those without actionable mutations currently may still be of therapeutic value and potentially benefit from targeted therapies in the future. In general, our findings provide new insights into patients with oligometastatic NSCLC who possess actionable molecular alterations and receive appropriately matched therapy, and lay the foundation for future prospective clinical trials of oligometastatic NSCLC guided by molecular profiling.
Aside from targeted therapy, immunotherapy has also expanded the therapeutic repertoire for NSCLC. Recent study has identified TMB as a promising biomarker for ICI treatment response [28]. We observed the median TMB of oligometastatic patients was 5.00 mutations/Mb. Alborelli et al. [54] enrolled 76 advanced NSCLC patients who had a median TMB of 7.00 (range, 0.00 to 22.00) mutations/Mb, which is close to our data in oligometastatic cohort. In this research, TP53 mutation, LRP1B mutation, SMARCA4 mutation, DDR genes mutations, and smoking were significantly positively correlated with TMB in oligometastatic NSCLC. These findings were consistent with previous studies showing that NSCLC patients with these genetic variations or biological risk factors were positively associated with a higher TMB [35,36,55–57]. In particular, this is the first time that the ABL1 gene has been found to be associated with higher TMB in NSCLC, especially in oligometastatic NSCLC. Therefore, specific concomitant mutations and distinct biological behaviors, such as smoking, may account for the higher TMB and help identify oligometastatic patients who benefit from immunotherapy, but more clinical studies are needed to confirm this relationship. Notably, statistical analysis didn’t indicate a significant association between TMB and sex, age, metastatic site, and mutations in CDKN2A, EGFR, RB1, CDKN2B, KRAS, PIK3CA, MET, MYC, and KEAP1. Actually, these clinical characteristics and some of these specific genes have been shown to be associated with TMB. For example, previous study reported that TMB had a better predictive power of immunotherapy response in women and was associated with increasing age, CDKN2A and EGFR mutations in lung cancer [31,33,34,58]. Future studies may recruit more oligometastatic patients to analyze the relationship between TMB and these clinical features or mutations.
To our knowledge, this study is the first to characterize the genomic alterations of oligometastatic NSCLC through NGS, which sheds light on the molecular characteristics and provides valuable biological and clinical insights into the intermediate state. We deeply analyzed the differences in somatic mutations and gene expression patterns between oligometastatic and polymetastatic tumors, as well as biological pathways that may play an important role in maintenance of oligometastatic status. Our findings may provide new insights into personalized treatment for oligometastatic NSCLC patients. The limitations of this study are worth mentioning. First, as this is a retrospective study, its database limits the investigation of other sources of potential bias. Second, in the absence of accurate diagnostic criteria, the diagnosis of oligometastasis was based on the current clinical consensus, so we may have created a confounding between patients with oligometastasis and those with polymetastasis. Thirdly, due to the great bias of the survival data in our research, the survival analysis was not performed. Fourthly, the imbalance of sample size between the oligometastatic and polymetastatic groups may result in statistical bias. Fifthly, the molecular mechanism related to the occurrence and development of oligometastasis found in this study is still lacking in further exploration and verification. Our findings should be validated in larger samples in future studies. In conclusion, our study comprehensively elucidates the unique mutational landscape of oligometastatic NSCLC patients and its differience from polymetastatic ones from the perspective of genomics, which may improve the selection of patients with real oligometastatic state and optimize personalized cancer therapy.
 XML Download
XML Download