

 PDF
PDF Citation
Citation Print
Print



Chronic liver disease and injury leads to regeneration of hepatocytes and fibrosis, characterized by the deposition of collagen in the extracellular matrix with replacement of hepatic parenchyma. It was initially thought that liver fibrosis with progression to cirrhosis was an irreversible process with subsequent mechanical effects (vascular remodeling, collateral circulation, portal hypertension), physiologic changes (retention of bile salts, coagulation abnormalities, metabolic disarray), and neoplastic implications (increased lifetime risk for developing hepatocellular carcinoma) [1,2]. This paradigm was initially challenged by Hans Popper in 1964 when he observed collagen resorption in idiopathic hemochromatosis patients after therapeutic phlebotomy [3,4]. In 1979, Perez-Tamayo [5] induced fibrosis and cirrhosis by chronic tetrachloride intoxication in animal models. Removal of the offending etiology caused near complete reversal of fibrosis with normal histologic findings [5].
The discussion continued to evolve until the next major breakthrough by Wanless et al. in 2000 [2], presenting serial liver biopsies from a patient with hepatitis B following antiviral treatment. They demonstrated chronological progression and reversal from cirrhosis to incomplete septal cirrhosis, correlating a decreasing viral load with clinical parameters [2]. Finally, the last two decades certified that successful treatment of chronic liver disease regardless of etiology (viral, biliary, vascular, steatotic, metabolic) caused fibrosis regression. Cirrhosis was once considered an endstage, irreversible process with orthotopic liver transplantation as an inevitable conclusion is now regarded as a dynamic, bidirectional process balancing fibrogenesis and fibrolysis [6,7].
PATHOBIOLOGY
With chronic and persistent liver injury, the liver responds by depositing extracellular matrix (ECM). This is characterized by type 1 and 3 collagen deposition in the portal tracts and lobules, and collagenous and non-collagenous ECM protein deposition in the space of Disse, which includes collagen type 3 and 4, laminin and fibronectin [8]. This wound healing response or scarring down of the liver begins when chronic liver injury causes apoptosis of hepatocytes leading to Kupffer cell activation and cytokine release: tumor necrosis factor (TNF), platelet derived growth factor (PDGF), and endothelin-1 (ET-1). Quiescent hepatic stellate cells (HSCs) in the space of Disse are normally inactive, dormant, fat storing cells, however, TNF activates stellate cells for transformation into myofibroblast like cells, depositing ECM as collagen types 1, 3, 4, and laminin. PDGF causes proliferation of stellate cells and ET-1 leads to contraction of stellate cells and vasoconstriction, affecting vascular resistance and liver blood flow. While normal sinusoidal spaces are lined by fenestrated endothelial cells, increased ECM material and collagen deposition closes sinusoidal endothelial cell fenestrations and the space of Disse, a process called capillarization of the sinusoids, preventing protein exchange between hepatocytes and flowing plasma [8-10].
Chronic portal and lobular inflammation causing hepatocellular necrosis with subsequent parenchymal extinction not only represents hepatocellular loss but an alteration in the surrounding microvasculature as chronic inflammation causes thrombosis of small branches of the portal vein, hepatic vein, and hepatic artery. This ischemic injury leads to subsequent cycles of parenchymal loss with compounding vascular compromise and remodeling, bile ductular reaction, and collapse of the hepatocellular trabeculae. When chronic injury persists, there can be years of parenchymal, architectural, and vascular remodeling, the latter which may be irreversible [2,11,12].
In contrast, matrix metalloproteinases (MMPs) and tissue inhibitor of metalloproteinases (TIMPs) are enzymes responsible for breaking down the extracellular matrix during fibrosis regression. There are approximately nine MMPs involved in the reversal of fibrosis with varying substrate specificity. These proteases are subcategorized into the following groups: stromelysins that break down protein substrates (i.e., laminin, gelatin, and type IV collagen), collagenases that degrade collagen (i.e., type III and I collagen), and gelatinases/type IV collagenases. MMPs are balanced and regulated by TIMPs, which irreversibly bind to and inhibit MMPs and are released by HSCs and Kupffer cells [13-15].
Retinoids (vitamin A) keep stellate cells in the quiescent state and prevent conversion to an activated state, inhibiting further differentiation into a myofibroblast-like phenotype. In tissue culture, activated HSCs show loss of retinoids, however, adding retinoids to the culture medium decreased type 1 collagen synthesis and deposition. Experimental studies showed that exogenous retinoid administration reduced expression of inflammatory cytokines such as TNF-α, maintained HSCs in a quiescent state and prevented Kupffer cells from releasing fibrogenic cytokines. Thus, there is an ongoing balance between fibrogenesis and fibrolysis involving an interplay of numerous factors during this dynamic, bidirectional process [16,17].
Go to : 
PATHOLOGIC FEATURES
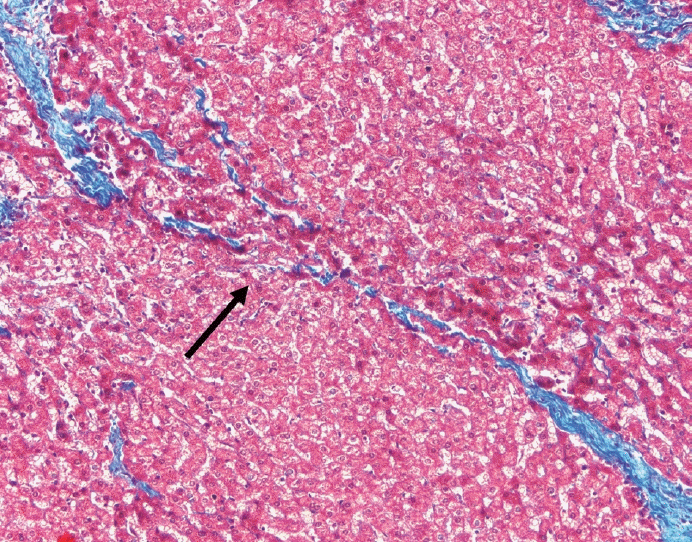
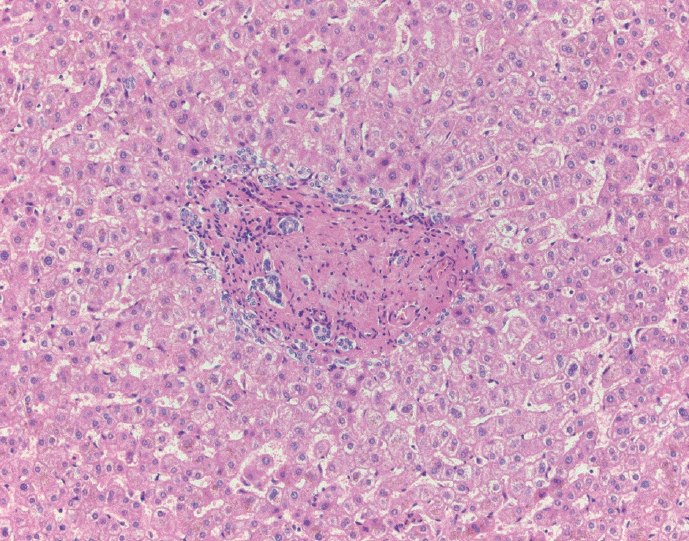
The typical cirrhotic nodule is surrounded by broad, thick fibrous septae with scattered chronic inflammation and bile ductular reaction encapsulating a nodule of regenerating hepatocytes. The central veins may not be visualized because they have been compressed and undergo thrombosis. As these outflow vessels are obliterated, small, collateral vascular channels develop and act as shunts to offset the increase in portal pressure. In contrast, fibrosis regression is comprised of three components, fragmentation and regression of the scar, vascular remodeling/distortion, and parenchymal regeneration. Inflammation subsides as the balance is shifted towards fibrolysis and fibrous septa become progressively thinner and wispier (Figs. 1–3). Eventually, hepatocytes push into or split the fibrous septa causing small perforations and fragmenting the septa (Figs. 4, 5). As the liver reverses bridging fibrosis and cirrhosis, delicate periportal fibrous spikes are left behind in the portal tracts [18,19].
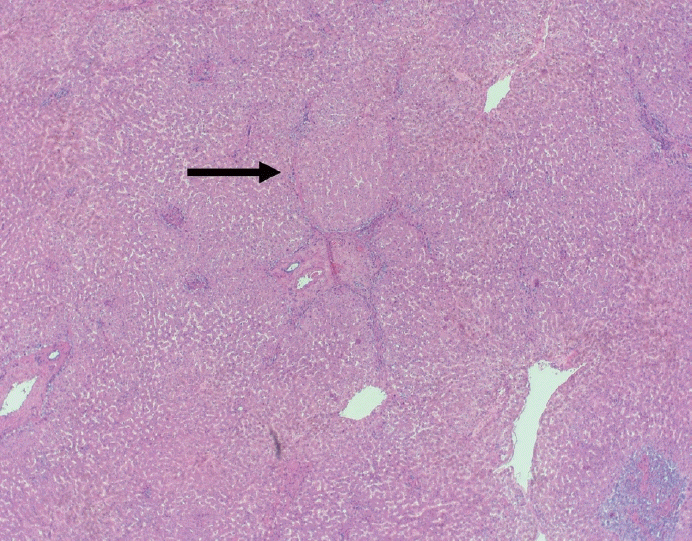 | Fig. 1.On low power, a nodular architecture is apparent in fibrosis regression (arrow). Fibrous septae are thin, incomplete and interrupted.
|
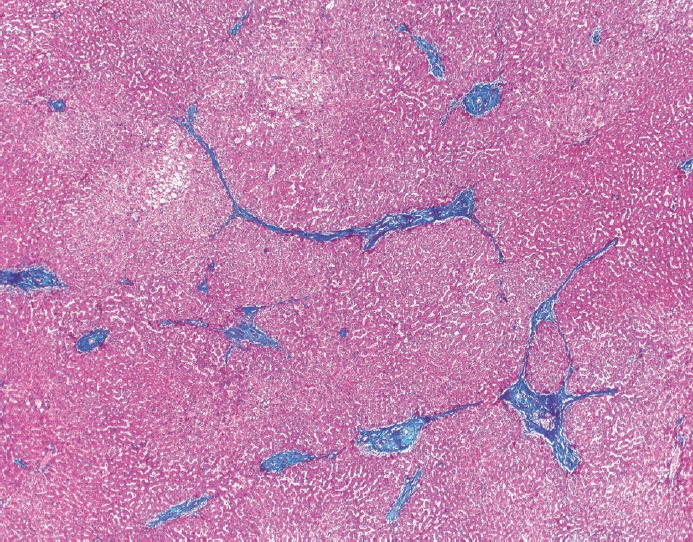 | Fig. .Trichrome stain highlights fibrosis regression with thin,
wispy, incomplete fibrous septa in a once cirrhotic liver.
|
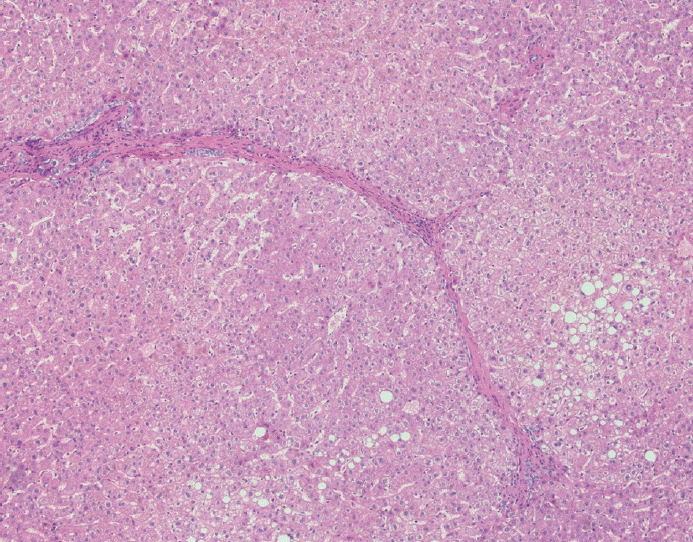 | Fig. 3.Fibrous septae are thin and wispy with a paucity of chronic inflammation. There is minimal, patchy bile ductular proliferation.
|
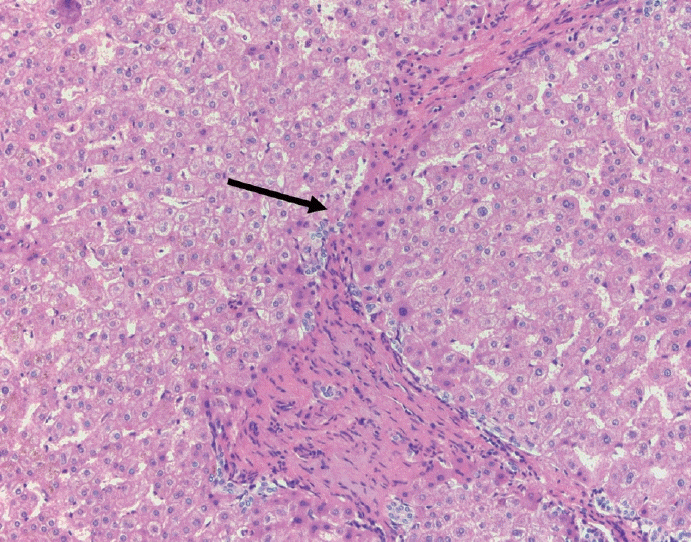
The portal tracts also demonstrate fibrosis regression as collagen is broken down, often leaving behind a paired bile duct and hepatic artery while the previously obliterated portal vein is not restored or visualized. This is known as a portal tract remnant. Hepatocytes prolapse or push into the fibroconnective tissue boundaries of the portal tract and are seen directly adjacent to the paired bile duct and artery (Figs. 6, 7). As the inflammation and hepatocellular extinction subsides, parenchymal regeneration begins, sinusoidal collagen is resorbed and hepatic trabeculae restores its normal architecture [18,20,21].
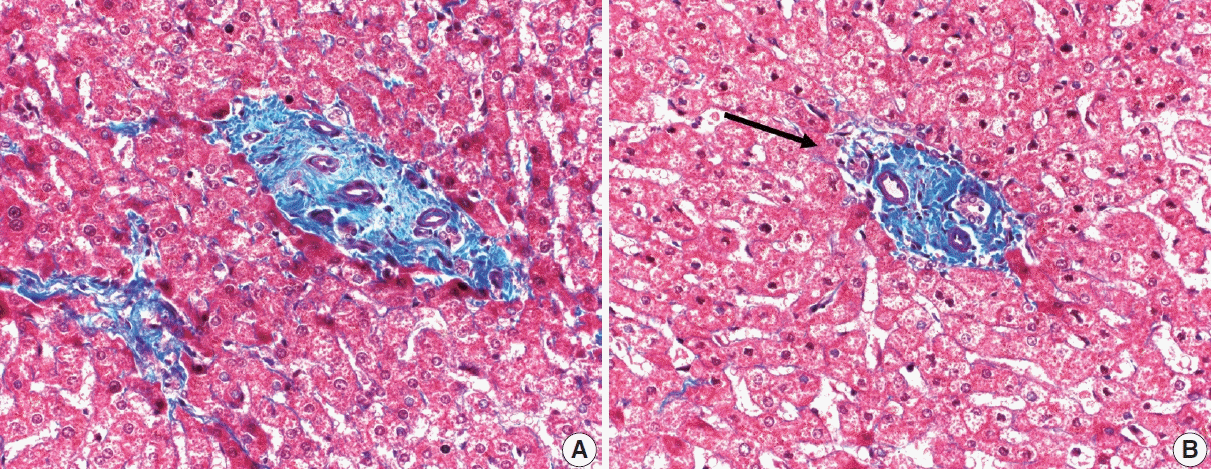
Go to :
CLASSIFICATION SYSTEMS
Traditional classification staging systems (i.e., Batts-Ludwig, Ishak, Metavir) characterized the architectural changes of fibrosis and cirrhosis in a linear pattern from no fibrosis to portal expansion, followed by step-wise periportal fibrosis, bridging fibrosis and ultimately cirrhosis (Table 1) [22,23]. Kutami et al. [24] introduced the Laennec classification system in 2000, which subdivided stage 4 cirrhosis into three categories: 4A, 4B, and 4C. Stage 4A was deemed mild cirrhosis with thin fibrous septa and large nodules, 4B with at least two broad septa but small nodules and stage 4C with large, broad fibrous septa with several small nodules. While this system is easily reproducible, there has not been widespread consensus in adopting this classification scheme [24].
Table 1.
Batts-Ludwig classification system for staging fibrosis
![]()
Due to effective treatments for chronic liver disease and the consideration of fibrosis and cirrhosis regression as a dynamic, bidirectional two-way street, Thiese et al. [18] proposed a new classification system for grading and staging hepatitis patients, the Beijing classification (Table 2). This system was proposed for the assessment of chronic viral hepatitis but has proven useful in describing activity and fibrosis for other etiologies of chronic hepatitis. It simplified grading activity and staging fibrosis and added a new, third category for determining the quality of fibrosis. Each biopsy is assessed a P-I-R score, predominantly progressive, indeterminate, or predominantly regressive as the major pattern of fibrosis. Predominantly progressive features show broad, thick fibrous septa with chronic inflammation, ductular reaction, parenchymal extinction, and congestion. A regressive pattern demonstrates wispy, thin fibrous septa with fragmentation, perforation, little or no inflammation and prolapsing hepatocytes into the portal tract as described earlier. Indeterminate denotes that the surgical pathologist is unable to distinctly classify a biopsy as P or R. The main benefit is that post-treatment biopsies can remain in the same stage (i.e., traditional stage 4 cirrhosis), but show significant changes in fibrosis quality (i.e., progressive to regressive or indeterminate), providing valuable prognostic information and determining response to therapy [18].
Table 2.
Beijing classification system
![]()
Besides a P-I-R score, liver biopsies are also graded and staged in the Beijing classification system (Table 2). Necroinflammation or hepatitis is graded as inactive (only portal inflammation or rare interface or lobular activity), non-severe active (variable interface and lobular hepatitis), and severe active (confluent necrosis, perivenular or bridging necrosis). Fibrosis is staged as early (no fibrosis, portal fibrosis), intermediate (focal or frequent fibrous septa, bridging fibrosis) and advanced (fibrous septa with focal or diffuse nodularity) [18]. The similarities to prior adopted grading and staging classification systems are evident. This new system for microscopic evaluation of grading and staging is susceptible to intra and interobserver variability, therefore, simplifying and decreasing the number of subcategories in grading activity and staging fibrosis improves reproducibility. Interobserver agreement between pathologists adopting the P-I-R staging system was high with a Kappa value of 0.71 (substantial agreement) [18,25]. The P-I-R system is also a valuable prognostic marker independent of the grade and stage, providing a snapshot for the current state of disease that strongly correlates with hepatic venous wedge pressures (hepatic venous pressure gradient [HVPG]) and portal hypertension [26]. In patients with chronic viral hepatitis, P, I, or R was an accurate surrogate marker for clinical outcome as successful eradication and clearance of hepatotropic viruses were predominantly R and unsuccessful treatments were predominantly P or I [18].
Go to :
CLINICAL IMPLICATIONS
In advanced liver disease, there is increased resistance to sinusoidal blood flow which causes portal hypertension or increased portal pressures. The gold standard for measuring portal hypertension is the HVPG and a gradient of less than or equal to 5 is within normal limits. Cirrhotic patients vary widely in their clinical presentation because the severity of cirrhosis ranges from compensated and asymptomatic to decompensated cirrhosis with ascites, esophageal varices, and hepatic encephalopathy. HVPG is an accurate prognostic marker in cirrhotic patients that risk stratifies the likelihood of those complications. Clinical studies show that with cirrhosis and fibrosis regression, there is a decrease in HVPG and portal hypertension-related complications with improvements in liver function and survival rates. A comprehensive assessment is based on clinical, hemodynamic (i.e., HVPG), and histopathologic features [27-29].
There are multiple approaches to achieving reversal of fibrosis and cirrhosis. The most common method is to control or cure the primary, underlying disease. This has a proven and successful track record for hepatotropic viruses (i.e., hepatitis B and C), autoimmune hepatitis, hereditary hemochromatosis, Wilson’s disease, and fatty liver disease. Anti-fibrotic agents target different steps in the pathobiology of fibrosis. These therapeutic drugs are focused on receptor-ligand interactions to prevent quiescent HSCs from transforming into activated HSCs, preventing the cascade of events that lead to deposition of ECM by inhibiting fibrogenesis, or accentuating the resolution of fibrosis through apoptosis or increased matrix degradation. There are over 500 active, clinical trials in this area of research [30].
Cirrhosis is also a major risk factor for developing hepatocellular carcinoma (HCC). With regression, there are physiologic and mechanical/pressure improvements in the patient’s condition, however, is there a reduction in the risk for developing HCC? There are two main factors that contribute to the pathobiology of HCC, the cumulative mutations of liver disease etiology (i.e., viral, metabolic, fatty liver, etc.) that form a clonal population/neoplasm and the surrounding extracellular matrix/ tumor microenvironment (TME) which consists of vascular abnormalities/remodeling and the fibrous stroma [31- 33]. Even when the initial insult is removed (i.e., hepatotropic virus cleared by medication, weight loss or medication for steatohepatitis, phlebotomy for hemochromatosis, etc.) and fibrosis regression is visualized, the driver mutations within hepatocytes persist. For example, in hepatitis B, there is DNA integration into the host genome leading to genomic instability, alterations to tumor suppressor genes, and TP53 mutations [34,35]. In non-alcoholic fatty liver disease, there are a different set of cumulative mutations, alterations in fatty acid beta-oxidation and insulin resistance [36]. As for the TME, vascular remodeling, ischemic injury, thrombosis of small blood vessels, lead to cycles of parenchymal extinction and regeneration with increased vascular endothelial growth factor expression and further genomic instability. Therefore, despite fibrosis regression, the risk for developing HCC remains high compared to the normal population and patients should continue to be actively screened and followed [32].
Go to :
CONCLUSION
Cirrhosis was once thought to be an irreversible process of endstage liver disease. This is no longer the case with treatment options for most chronic liver diseases. Fibrosis regression is characterized by thinning of the fibrous septa with hepatocytes pushing into the septa and eventual perforation. This leads to periportal spiking within portal tracts and prolapsed hepatocytes into the boundaries of the portal tract stroma. Obliterated portal veins during progressive fibrosis and cirrhosis due to parenchymal extinction, vascular remodeling, and thrombosis often leaves behind a bile duct and hepatic artery within the portal tract. To characterize these histopathologic features, the Beijing classification system offers an accurate snapshot of a dynamic process between fibrogenesis and fibrolysis. Even with fibrosis regression, vascular lesions/remodeling, parenchymal extinction, and cumulative mutational burden persists and patients should continue to undergo clinical surveillance.
Go to :
 XML Download
XML Download