

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Neurofibromatosis type 1 (NF1) is an autosomal dominant genetic disorder involving multiple organs, with variable phenotypes. It develops sporadically without a family history in approximately half of the patients [1]. Central nervous system vasculopathy in patients with NF1 is associated with diverse clinical manifestations. Cerebral arteriopathies, including steno-occlusion, ectasia, and aneurysmal changes, have been reported in the literature, but their prevalence has not been estimated [1,2].
Not only is it rare to encounter moyamoya-like vessel changes in patients with NF1, but it is also exceptional to observe concomitant arteriovenous malformations (AVMs) in the remaining infratentorial vasculature. Herein, we report a case of contradictory occlusive and excessive intracranial vasculopathy in a single patient with NF1.
CASE REPORT
A 28-year-old man visited our neurological clinic with involuntary movements of his right arm which lasted for 3 months. He described oscillating rhythmic right hand tremors. It started after he survived the rupture of an ipsilateral cerebellar AVM involving the cerebellopontine area. His medical history revealed that he had NF1 since he was 10 years old. His diagnosis was supported by multiple hyperpigmented brownish macules measuring >15 mm (café-au-lait spots), and axillary/inguinal freckles. He endured one or two episodes of infrequent afebrile epileptic seizures per year until the age of 9, which regressed spontaneously. His intellectual ability lagged slightly behind that of his peers and he manifested an attentional deficit without social incompetence. His motor development was not delayed; however, he appeared to be shorter than his age-average height. At 28 years old, his size reached 164 cm.
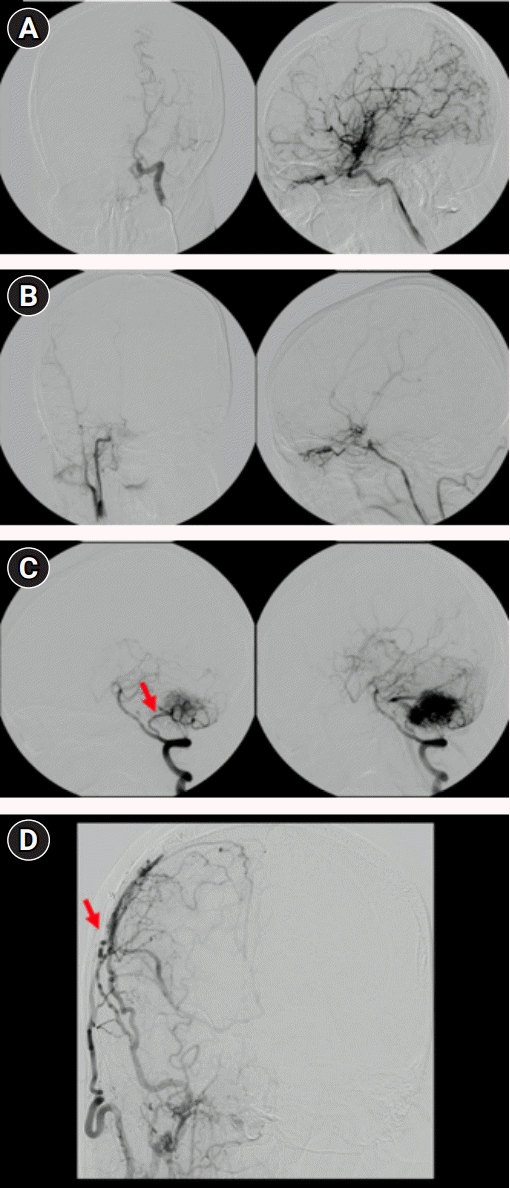
At the age of 15, 13 years before the AVM rupture, he reported clumsiness and awkwardness of left limb movements. He was diagnosed with asymmetric moyamoya syndrome (Suzuki grades III and VI in the left and right hemispheres, respectively (Fig. 1A and B) and right cerebellar AVM. The right posterior inferior cerebellar artery (PICA) mainly supplied the vascular tuft, which drained into the sigmoid sinus (Fig. 1C). A Suzuki grade VI in the right hemisphere was presumed to be the cause of the patient’s symptoms. Right superficial temporal artery to middle cerebral artery bypass was performed to revascularize the right hemispheric vasculature. The infratentorial AVM was not intervened at the time. After the surgery, the patient stopped experiencing left appendicular symptoms. The bypass conduit was maintained for 13 years (Fig. 1D).
Neither his parents nor brother were diagnosed with any neurological disease. The patient was not taking any medications that might have provoked his unilateral tremor. Right upper and lower limb ataxia were observed on neurological examination. The motor power of the extremities was graded as the Medical Research Council 5. The patient was unstable on foot and required assistance while walking. His right hand tremors had a rhythmic supination-pronation nature within 2–4 Hz when his hand rested in the mid-position between supination and pronation. The Mini-Mental Status Examination score was 23/30, revealing global cognitive impairment. However, he had the total capacity to engage in conversations and respond to commands. Serological assessment, including copper metabolism, revealed no metabolic derangement relevant to the tremor. Ophthalmological evaluation did not reveal any copper deposits.
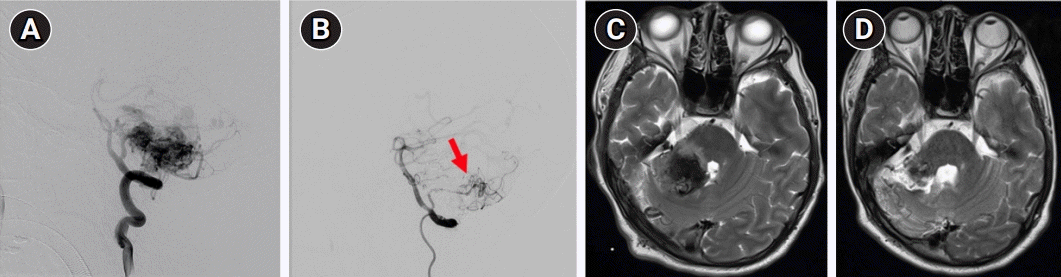
Six months before the tremors, the right cerebellar AVM had ruptured. The right anterior and posterior inferior cerebellar arteries (AICA and PICA) and superior cerebellar artery supplied the AVM. The AICA and superior cerebellar artery feeders were successfully embolized, whereas the PICA was partially blocked with remaining residual flow (Fig. 2A and B). The extant PICA was not intervened further because its flow velocity significantly decreased and other feeders were blocked. Regular follow-ups were planned to monitor for any changes. Brain magnetic resonance imaging at rupture and at follow-up (Fig. 2C and D) revealed edematous hemorrhage in the cerebellum/cerebellar peduncles and incomplete regression, respectively.
After 6 months of follow-up, his position-specific tremors persisted despite improvement in his limb ataxia. The patient could walk independently with his feet wide. Although his tremors did not disappear, they did not interfere with his daily activities of living, and ataxia rehabilitation was maintained without any medication.
DISCUSSION
NF1 is a multisystem disease with diverse progression and severity throughout life [3]. Phenotypic variants occur across multiple organs and represent structural and/or functional deficits [3-6]. Its diagnosis can be postponed when an affected individual without a known family history presents only with skin hyperpigmentation and later develops other signs that fulfill the diagnostic criteria [1,3]. The patient’s diagnosis of NF1 was delayed at 10 years of age because other non-skin manifestations, such as intellectual/attentional deficits and below-average stature, became apparent later and indicated its suspicion.
Five years after his diagnosis, asymmetric moyamoya syndrome was identified, and the symptomatic hemispheric vasculature was rescued by bypass surgery. Although cerebral arteriopathy is rarely reported, NF1 is associated with cerebrovascular abnormalities in the adult and pediatric populations [1]. Moyamoya syndrome is one of its subtypes with higher risk of ischemic or hemorrhagic stroke than in the general population [2,7]. Right hemispheric ischemia caused by asymmetric steno-occlusive intracranial cerebral vessels (more severe on the right) was interpreted as evoking left appendicular awkwardness when demanding execution was required. The patient's infrequent epileptic seizures, which resolved spontaneously, could also be explained by cerebral vasculopathy. Hemodynamic insufficiency could have occasionally kindled epileptogenic firing above the threshold, which disappeared after rescue surgery.
Ecstatic or aneurysmal vasculopathy associated with NF1 has been investigated in a systemic review, but infratentorial AVM has seldom been reported, let alone concomitant cerebrovascular abnormalities in a single NF1 individual [2-4,8,9]. The pathomechanism underlying cerebrovascular disease is poorly understood [1]. The coexistence of opposing vasculopathies (occlusive vs. hyperplastic) may be a coincidence. However, simultaneous observations of advanced moyamoya syndrome and a large infratentorial AVM in childhood, which affected approximately three-quarters of all intracranial vessels, suggested that the chronic pathobiology of NF1 took place for a prolonged time and played a role in its vasculopathy.
The position-specific tremor associated with limb ataxia was ipsilateral to the AVM rupture. The exact pathomechanism remains elusive, but its time-locked appearance after hemorrhage implies that the destruction of a central oscillator network, such as the Guillain-Mollaret triangle, may have caused its hyperkinetic movements [10]. Cerebral vasculopathy is a consistent and relevant manifestation of NF1 and can take variable forms with extensive involvement, even in early childhood, as seen in this case report. Its prevalence and natural history are unknown because active screening for cerebrovascular diseases is not routinely performed [2]. Approximately 44.5% of NF1 patients who demonstrated vascular abnormalities were asymptomatic, emphasizing the need for early screening and not waiting for symptoms to emerge [2].
In summary, we report a case of chronic NF1 with extensive concomitant opposing cerebral vasculopathy. Investigations and interventions for cerebrovascular disease were only performed when the patient became symptomatic. The overall annual rate of AVM rupture is estimated to be 2.3% (1.3% for unruptured vs. 4.8% for ruptured AVM), and specific genes increase the risk of rupture [11,12]. Therefore, as in this case report, AVMs with genetic predispositions require active surveillance with a tailored prediction of rupture and preemptive intervention. Extensive cerebral vasculopathy can occur even in childhood and is life-threatening if not adequately addressed.
 XML Download
XML Download