

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
The incidence of status epilepticus (SE) ranges from approximately 5 to 40 per 100,000, based on several population-based studies across the US [1,2], and the incidence of super-refractory status epilepticus (SRSE) is 0.7 per 100,000. The response to treatment, its evolution time and duration, and the need to use one or more antiseizure drugs define SE as refractory or super-refractory. In 2015, the Task Force of the International League Against Epilepsy defined SE as a prolonged seizure that exceeded the intrinsic termination mechanisms at a certain point in time from the onset of the seizure (t1) and persisted beyond a second point in time (t2), the latter being the time at which cortical damage occurs. It also defines refractory status epilepticus (RSE) as one that persists beyond t2 time point despite treatment with first- and second line antiseizure drugs. It also defines SRSE as an SE that persists for more than 24 hours after the addition of a third-line drug, generally an intravenous anesthetic [1].
These definitions and their determinant aspects have clinical and therapeutic implications and dimension the consequences of the status, which forces the implementation of treatment with a certain "aggressiveness" and is a quick start to try to limit neuronal damage and improve neurological and clinical results [2-4]. The incidence of refractory and SRSE is approximately 40% among patients presenting with SE, depending on the cause, with mortality approaching 50% [5].
Ketamine, a drug derived from phencyclidine, was synthesized in 1960 and reached peak popularity in the 1970s. In 1982 it was reported that it could have another possible mechanism of action by agonism of µ, δ, and ɣ opioid receptors [6]. Another possible mechanism of action is the inhibition of monoamine oxidase, which prolongs the half-lives of serotonin, dopamine, and norepinephrine [7,8]. In a study published in 2011, a decrease in the production of interleukin (IL)-6, IL-8, IL-10, and tumor necrosis factor-alpha was found in patients undergoing cardiopulmonary surgery who received ketamine, which gives it certain anti-inflammatory properties [9]. Another study in 2015 observed that it caused a decrease in calcium concentration at the cytosolic level in cardiac myocytes, and thus counteracted its effect in states of hypoxia, ischemia, oxidative stress, and hypertrophic cardiomyopathy [10]. Ketamine occurs in two forms: the S isomer, which is three times more active than its opposite, the R-enantiomer. The most widely used pharmacological presentation is a racemic mixture of both molecular forms [11-13]. Despite its widespread use as an anesthetic during surgical procedures, it was not until the 1990s that an article was published showing its use in treating SE [14,15].
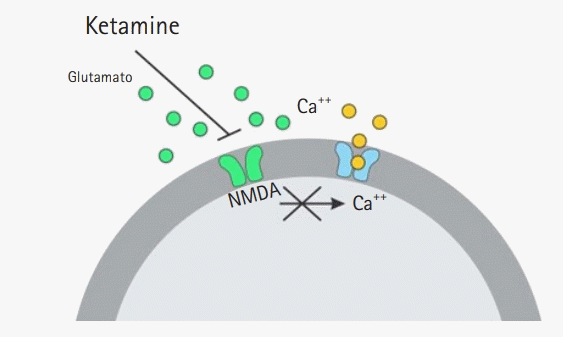
Glutamate receptors (pharmacological targets of ketamine) are classified into two types: metabotropic receptors (mGluRs), which promote the activation of second messengers via the activation of transmembrane G proteins, and ionotropic receptors, which are coupled to an ion channel and allow the entry of various ions, mainly calcium [16,17] (Fig. 1). Ionotropic receptors are divided according to the affinity of their specific agonists; here, N-methyl-D-aspartate (NMDA) receptors belong to this family.
Excitotoxicity is characterized by neuronal death induced by the excessive release of glutamate and overactivation of its receptors [18]. This event is associated with various disease states of the central nervous system, including epilepsy, hypoxia, ischemia and trauma. This overstimulation increases intracellular calcium concentrations; promotes lipid peroxidation of the cytoplasmic membrane, endoplasmic reticulum, and mitochondria and causes cell death [19,20].
METHODS
This scoping review seeks to synthesize available information from different domains of drug use in SRSE, specifically ketamine. No specific questions were raised and the definitions and variables used in different publications on the use of ketamine in SE were explored. Two researchers searched for articles using keywords in the PubMed, Embase, SciELO, Bireme, Latindex, and Google Scholar databases, and medRxiv was explored for gray literature. The words that were used to construct the search strategy were “status epilepticus,” “refractory,” and “ketamine.”
RESULTS
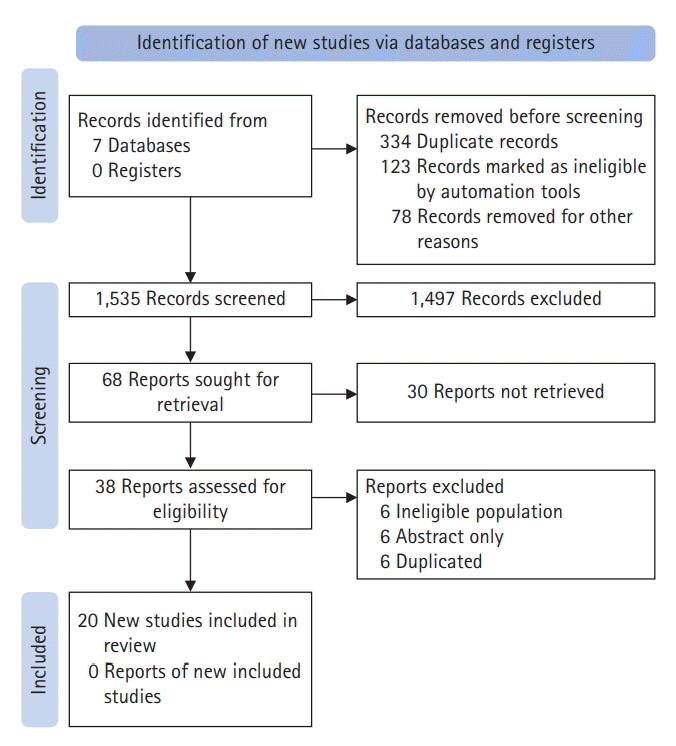
A search of seven bibliographic databases identified 38 articles related to the terms of interest published between 1990 and 2022. Articles in English or Spanish were selected based on their titles or abstracts. After filtering by content and relevance, 20 articles were selected for the final document construction (Fig. 2). The extraction results were synthesized and presented as a qualitative description of the information and synthesis in quantitative data tables.
Pharmacokinetic profiles of ketamine
Table 1 shows the main causes of SE, including head trauma, cerebrovascular disease, infections, tumors, autoimmune disorders, and hypoxic-ischemic encephalopathy. The etiology of SE and SRSE is particular and suggests that both conditions are more likely to develop due to acute neurological damage associated with a persistent inflammatory state, but not specifically with the presence of epilepsy, an underlying pathology.
To initiate the pharmacological treatment of SE, it is crucial to understand the mechanism by which seizures become refractory. Although the molecular pathophysiology of SE is complex, it complies with some of the body's “laws of homeostasis.” For example, seizures are perpetuated by an imbalance between excitatory and inhibitory mechanisms in neuronal tissues [21]. γ-Aminobutyric acid (GABA) is the primary inhibitory and Glutamate is the main excitatory neurotransmitter that mediates excitation by stimulating the NMDA receptor [22,23].
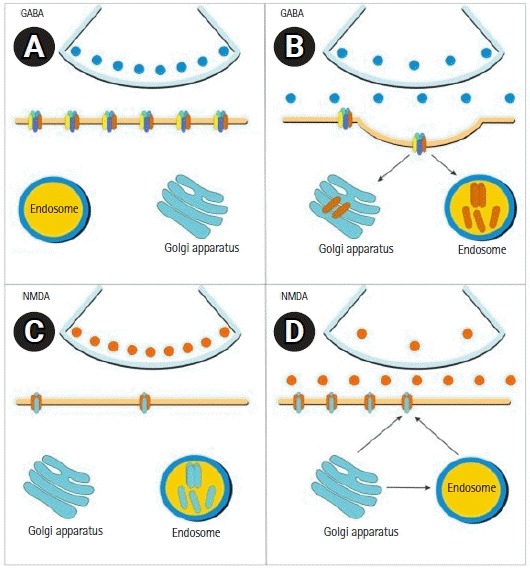
After several minutes of constant seizure activity, “receptor trafficking” appears. This is a phenomenon in which GABA receptors decrease through a process of “internalization,” generating a reduction in GABAergic activity. The number of glutaminergic receptors on the cell surface increases due to “externalization” from the cytosol to the cell membrane, producing increased neuronal excitatory activity [24-27]. This is clinically reflected in rapidly progressive resistance to benzodiazepines [28]. In this scenario of impaired GABAergic activity and increased excitotoxicity, NMDA receptor antagonists, such as ketamine, become therapeutically relevant because they have a pathophysiological substrate that favors their effectiveness (Fig. 3).
Other events are believed to occur at the tissue and cellular levels and contribute to the development of RSE and SRSE. Among these events, there may be mitochondrial failure or insufficiency [29] and activation of the inflammatory cascade [30,31], which make the blood-brain barrier vulnerable together [32,33]. In treating SE, the primary therapeutic objective is to rapidly control ictal activity to limit neuronal death mediated by excitotoxicity and reduce the systemic complications generated, with benzodiazepines considered the first-line treatment. Other anticonvulsants such as phenytoin, levetiracetam, and valproic acid are the most widely used second-line drugs [34,35].
Having explored the pathophysiological substrate for which a third-line drug with effects on neuronal excitatory activity, specifically on NMDA receptors, could play a leading role, it is time to study the potential role of ketamine. In a retrospective study published in 1996 by Walker et al. [14], a case of SE was reported in which ketamine was used as an anticonvulsant drug, demonstrating a favorable therapeutic response, and no noteworthy adverse effects related to its use were reported. Since then, there have been 20 publications on ketamine and SE: nine case reports, eight retrospective case series two systematic reviews, and one observational (Table 2) [5,14,15,36-52].
Usefulness of ketamine in RSE
In the analysis of the obtained information, two questions arose that motivated this investigation. Is ketamine useful in the treatment of RSE? Although seizures at any given moment may have a certain degree of clinical subjectivity, the two parameters used in the publications to determine the effectiveness of ketamine in RSE were clinical and electroencephalographic cessation of epileptiform activity. Between case reports and case series, there are 265 patients. A systematic review conducted in 2014 [46] grouped 110 patients, and another conducted in 2018 grouped 289 patients [5]. Of the 19 case studies, 12 (63%) achieved complete seizure control, whereas in the remaining case studies, seizure control was partial (31%) or absent (5%).
Table 2 contains some information from the results of studies on using ketamine in RSE. Each publication details, for example, the route of drug administration (intravenous or oral), the administration or not of loading dose, and the response to treatment, which was classified into three categories: good response, referring to patients in whom the administration of ketamine ceased seizure activity; partial response, referring both to patients in whom seizures recurred after administration of the drug and to groups of patients where seizures were controlled in some patients and not in others; and no response, referring to patients in whom ketamine treatment had no effect on the duration of treatment expressed in hours, days, and months, and adverse drug reactions [5].
In more than 70% of studies regarding seizures controlled with ketamine during the first 48 hours of treatment, it is evident that this drug is effective in treating seizure activity in the RES and that its therapeutic effect is achieved within a few hours [5]. Its effect appears to be related to early onset and is also likely to have a direct relationship with the administered dose, with reported doses being higher than 0.9 mg/kg/hr [45,50,51].
A negative aspect that limits the benefit of ketamine in this clinical scenario is recurrence after its cessation. This recurrence appears in 10%–60% of cases in which seizures are successfully suppressed [50,52]. The recurrence of seizures when withdrawing ketamine treatment depends on multiple factors that are difficult to associate with each other, including the etiology of RSE, comorbidities, severity of the convulsive condition, medications used concomitantly or previously, and disorders of the internal environment. This disparity makes it difficult to establish a causal relationship between these two events.
• What is the pharmacodynamic profile of ketamine in RSE? We will analyze the information found on the loading dose, maintenance dose, start time of the drug, duration of treatment, and its therapeutic and adverse effects.
In 70% of the studies, a loading dose of ketamine was administered at a rate of 0.5 to 5 mg/kg. The intravenous infusion doses ranged from 0.05 to 10.5 mg/kg/hr, and for patients who received the drug enterally, the doses ranged from 50 to 250 mg twice a day. Ketamine treatment was administered between the first 24 hours and 140 days after the diagnosis of RES. The duration of ketamine treatment ranged from 2 hours to 29 days. The electroencephalographic response to ketamine treatment, as determined by the burst-suppression phenomenon, was as rapid as 2 hours or as delayed as 28 days.
Adverse effects of ketamine
The reported adverse effects included transient arterial hypertension (one case), supraventricular tachyarrhythmia (two cases), cerebellar syndrome (one case), metabolic acidosis in co-administration with midazolam (one case), and cardiovascular collapse associated with metabolic acidosis (two cases) [5]. However, the heterogeneity in pharmacodynamic information found when using ketamine as a third-line drug in RSE [5] constitutes an unfavorable aspect in determining its therapeutic index and other aspects of its pharmacodynamics. Little information is available regarding the oral administration of the drug. Oral ketamine has been used to control chronic pain in patients with neuropathic pain, cancers of different types, trigeminal neuralgia, and phantom limbs, among other conditions. The analgesic effect is mainly based on NMDA receptor activity. Although the bioavailability of orally administered ketamine is only 16%, its major active metabolite, norketamine, retains its NMDA receptor-antagonistic properties and has less affinity for NMDA receptors.
Until now, it has not been possible to determine the appropriate dosage because the therapeutic range is extensive and ranges between 45 and 1,000 mg/day. However, in patients treated with enteral ketamine, its effectiveness in seizure control appears to be maintained, thus constituting an alternative in cases where the intravenous route cannot be used [13,41]. Regarding adverse effects, it is not easy to establish the association between these and ketamine. Is not a first- or second-line drug, once treatment with this NMDA antagonist is initiated, it is difficult to assign any adverse reactions to ketamine in a critically ill, polymedicated patient with at least two centrally acting drugs. Additionally, the wide range of doses administered and treatment durations complicate the establishment of the causality of a clinical condition with a particular drug. However, several publications have reported sialorrhea, hepatotoxicity, cholestasis, cardiac arrhythmias, and metabolic acidosis related to ketamine use [37,39,45,53-55]. Although this drug is popular because it does not have hemodynamic depressant effects, this is probably due to the release of endogenous catecholamines, which increase peripheral vascular resistance and heart rate [56-58]. Paradoxically, some reports indicated that the effects of endogenous catecholamines on intracellular calcium currents could prolong cardiac action potential and have adverse inotropic effects [59-61]. In patients undergoing multimodal neurological monitoring, ketamine infusion did not generate harmful changes in intracranial pressure, and it was possible to reduce vasopressor requirements [50].
This particularity of the cardiovascular system may correlate with the cardiovascular collapse observed in two cases where ketamine was used [5]. Metabolic acidosis, which has also been reported in the treatment of RSE, has no clear cause. Some hypotheses suggest that hydrochloric acid, a diluent of midazolam (the first-line drug), can cause hyperchloremia, and thus contribute to acidosis, homeostatic imbalance, and cardiovascular collapse [5].
DISCUSSION
The main goals of RSE and SRSE treatment are to preserve cortical function and reduce morbidity and mortality related to neuronal damage caused by prolonged seizure activity. Cognitive, behavioral, and functional alterations have been reported at rates higher than 75% in patients with RSE and are clearly related to the duration of seizures [62,63]. Given the current knowledge about the biomolecular changes that occur in the neuronal membrane during prolonged seizures and the early onset of drug resistance to GABAergic drugs, there are clear reasons to consider the early initiation of ketamine treatment in RSE protocols [3-26,28-35,39,41,46].
The scant prominence of ketamine as a rescue treatment for RSE is attributable, in some way, to medical inexperience with the use of the drug in this context, the heterogeneity of the information available regarding its pharmacodynamic profile and side effects, and the lack of studies that include ketamine within their protocols. Although in most studies ketamine was not used in the initial hours of ER and was only used after various anticonvulsants, it was effective in controlling seizures when administered, usually within the first 12 hours [5]. However, there is currently insufficient scientific evidence to support the use of ketamine as a first-line drug or as a monotherapy in the management of RSE and SRSE.
It is reasonable to assume that there is fear and insecurity when using drugs with such disparate pharmacodynamic profiles. It is likely that, as has happened with other drugs, further studies will endorse its use, even more so in extreme clinical scenarios, such as supra-refractory status. For example, only after several years of intravenous lorazepam and diazepam could their toxicity be overcome, finding that it was due to propylene glycol, the excipient used in their pharmacological presentation, rather than this toxicity being inherent to these benzodiazepines. Accordingly, the statistical power of the studies we included was insufficient to relate the infusion of ketamine to a positive or negative impact on the mortality of these patients.
Few studies of report type and case series with small sample sizes and low statistical power, have examined the use of ketamine in RSE and SRSE. Despite the limited available evidence, the rapid efficacy of ketamine in the treatment of RSE and SRSE has been documented. This fact, in addition to the molecular and pathophysiological substrates that support the use of the drug, should encourage further research in this regard. One of the aspects to be clarified is the safety profile of ketamine and its pharmacological interactions with the other anticonvulsants used in RSE and SRSE treatment to elucidate the mechanisms of metabolic acidosis and other homeostatic alterations reported in the studies with ketamine and RSE.
CONCLUSIONS
The treatment of RSE and SRSE represents a therapeutic challenge because of their high mortality rates and poor neurological outcomes. Early initiation of anticonvulsant therapy and its effectiveness are crucial aspects of patient prognosis. Several animal models have shown that, in persistent seizures, the cell membrane experiences a decrease in GABAergic receptors and an increase in the expression of excitatory receptors, including NMDA glutamatergic receptors.
There are few studies on the use of ketamine in RSE and SRSE, and most of the information comes from case reports, case series, and retrospective studies, mostly with small or unique samples. Although the information is heterogeneous, it suggests that ketamine, used as a third-line drug for RSE and SRSE, is effective in controlling seizures. In this sense, it would be a drug with pathophysiological endorsement for use since the current evidence is favorable, if limited. Despite this, the nature of adverse reactions reported with the use of ketamine varies, and its cardiovascular effects and changes in the internal environment seem to be predominant. Given the methodological limitations of most publications in this regard, information on neurological outcomes measured using the Glasgow Outcome Scale (and modified Rankin scales in patients with SE and SRSE who received ketamine is heterogeneous and inconclusive.
The lack of randomized prospective studies constitutes a significant limitation in recommending and including ketamine in the RSE and SRSE protocols. Additionally, based on the changes in the biochemistry of the neuronal membrane and its receptors that occur in RSE, the question arises as to whether anti-glutaminergic drugs should be started early or as third-line drugs for this pathology. Despite these limitations, ketamine appears to have a promising outlook for RSE and SRSE.
 XML Download
XML Download