

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Traumatic brain injury (TBI) is one of the leading causes of death among young people worldwide. Furthermore, it is one of the most common causes of hospitalization [1]. TBI is a type of functional brain injury that can affect brain function in somatic, cognitive, and emotional domains. Headache is the most common symptom of TBI, followed by dizziness, balance disorders, loss of awareness of time and place, and sleep disorders [2,3]. TBI can be classified into two stages, with the primary phase as the initial hit that moves the brain inside the skull, and the secondary phase develops over time as a result of cellular events that cause additional damage and can last from a few days to a week or month [4,5]. TBI causes temporary membrane abnormalities that lead to ion and neurotransmitter redistribution, resulting in an increase in levels of calcium and stimulant amino acids, followed by the release of potassium, which inhibits neuronal activity. When sodium-potassium Na+/K+-ATPase pumps regain their balance, the need for energy increases while cerebral blood flow remains low. Disorders in autonomic nervous system regulation can last for several weeks, and the brain may be more vulnerable to damage [5]; however, evidence for drug treatment is limited. The goal of therapy is to maintain symptoms under control. Rest is the most important component of treatment because it can exacerbate concussion symptoms and prevent recovery [6]. Evidence suggests that autophagy increases after a TBI [7,8]. In stroke rats, the administration of a selective autophagy inhibitor (3-methyladenine) has been shown to have neuroprotective effects [9].
Chloroquine (CQ), an antimalarial drug, has been shown to easily cross the blood-brain barrier (BBB) and inhibit autophagy in a variety of disorders, including Alzheimer disease and brain ischemia [10,11]. CQ is also effective in treating cranial oxidative stress, as it reduces the reactive oxygen species in the brain. Therefore, CQ is regarded as neuroprotective in the brain following TBI [12]. CQ, an autophagy inhibitor, can be used to reduce brain tissue destruction and improve neurological function following TBI. Autophagy is activated after brain injury, which can result in neuronal and astrocytic injuries. Matrix metalloproteinases (MMPs) are a group of zinc-dependent endopeptidases that degrade the extracellular matrix (ECM) and other proteins. MMPs are required for ECM remodeling, wound healing, and tissue morphogenesis [13]. Under pathophysiological conditions, MMPs act as proteolytic enzymes that destroy the ECM and the BBB binding proteins. MMPs are secreted by astrocytes, neurons, epithelial cells, fibroblasts, and osteoblasts, and they interfere with natural physiological processes, such as angiogenesis, neurogenesis, inflammatory processes and apoptosis. MMP-2, MMP-3, and MMP-9 levels increase as a result of brain trauma, resulting in neuritis and cell death [14]. We investigated the effects of intraperitoneal (IP) CQ injection on neurological scores, cerebral edema, BBB permeability, and the amount of MMP-9 after inducing severe brain trauma using the Marmarou method [15].
METHODS
Animals and groups
A total of 120 male Wistar rats (250–330 g; Mazandaran University of Medical Science) were housed under standard conditions (air-conditioned room at 22 °C±2 °C temperature with a 12-hour light:12-hour dark cycle) with free access to clean water and food. Animals were injured using Marmarou free-fall TBI technique [16]. Rats were randomly divided into the following groups: (1) TBI group (TBI, n=24): rats were subjected to anesthesia, surgical opening, and TBI induction, (2) Saline group (vehicle, n=24): similar to TBI group, but an equal volume of the 0.9% normal saline (solvent of CQ) was injected intraperitoneally 30 minutes after TBI induction, (3) CQ groups (CQ 1.5, 3, and 6, n=24 in each group): CQ with different doses of 1.5, 3 and 6 mg/kg were injected IP 30 minutes after TBI induction [10,11].
Model of diffuse TBI
The animals were anesthetized using ketamine and xylazine, and the animals were intubated through the mouth (the tracheal cannula was connected to the breathing pump to control breathing and fend off hypoxia). Diffuse TBI was performed using the Marmarou method [17]. To distribute the effect and keep away from the cranium fracture, the scalp was incised to show the cranium, and a metallic disk with a thickness of 3-mm and 10-mm diameter was glued to the bone centrally alongside the coronal among bregma and lambda using polyacrylamide glue with a view to distribute the blow onto a larger area. Subsequently, the animal was positioned in a susceptible function on a 10-mm thick foam bed, and a 400-g blunt steel cylinder was dropped vertically through freefall from a height of 2 m. Following trauma induction, the body temperature was maintained via the methods at 37 °C by setting the animals on a heating pad. Following trauma induction, the rats were immediately connected to an animal breathing pump (TSE animal respirator, Germany). After recovery, the animals were disconnected from the pump and housed in individual cage [17-21].
BBB permeability assessment
The BBB’s permeability to small and massive molecules was assessed qualitatively using intravenous 2% Evans blue dye (molecular weight 6,900, 20 mg/kg). The animals were anesthetized 4 hours after trauma with thiopental (50 mg/kg IP), and 20 mg/kg Evans blue dye (1 mL/kg) was injected through the jugular vein (n=8 per group). One hour after injection, the thorax of the anesthetized animal was opened. The descending aorta was clipped, and isotonic heparinized saline solution (approximately 300 mL) was infused into the circulatory gadget through the left ventricle to remove the intravascular dye. Each jugular vein was then cut, and saline infusion was continued until the clean liquid strolled out of the jugular vein. The mind was changed to be eliminated immediately and homogenized after weighting. A 20 mL solution of acetone (14 mL)+1% sodium sulfate (6 mL) was infused into the homogenized mind and placed at the shaker for 24 hours. Then, 1 mL of trichloroacetic acid combined with 1 mL supernatant was placed at a fab place (20 °C) for 3 minutes. After centrifugation of the solution at 2,000 rpm for 10 minutes, Evans blue dye absorption from 1 mL supernatant was measured at 620 nm using a spectrophotometer (Pharmacia Biotech). The following system was used to calculate Evans blue dye content:
Determination of brain water content
Brain water content was assessed 24 hours after trauma induction using the dry-wet weight process. Briefly, the brains of anesthetized animals were removed and weighed to obtain the wet weight. The tissue was placed in an incubator at 60 °C–70 °C for 72 hours and reweighed to obtain tissue dry weight. Finally, the percentage of brain water content was calculated using the following equation:
100×(wet weight–dry weight)/(wet weight) [22].
Evaluation of neurological outcomes
Neurological function was evaluated using the veterinary coma scale (VCS) and expressed as a score from 3 to 15, which was comprised of three parts: motor, eye, and respiratory function. According to VCS criteria, higher scores mean superior neurological outcomes, and lower scores indicate a more severe neurological deficit. The detailed scoring system is shown in Table 1. In the present study, the VCS was measured 1 hours before trauma (Pre), immediately after trauma (D0), and 24 (D1), 48 (D2), and 72 (D3) hours post-trauma (n=8 per group) [23,24].
Vestibulomotor function evaluation
The beam task was used based on previous training to evaluate vestibulomotor function three times following the damage. Beam training was performed prior to TBI. For the duration of the training, rats were given five trials to traverse a 100 cm long beam with a width of 4 cm, and an additional five trials to travel a 100 cm long beam with a width of 1.5 cm. On the testing days, the mean of the three endeavors for each rat was recorded. The beam walk trial was started with the rat located at the beginning of the beam (2.5×100 cm) and ended when the animal effectively traversed a distance of 100 cm. A maximum duration of 60 seconds was permitted for each experiment. The beam balance trial started with the rat being placed on the 2 cm beam and the time spent balancing for 60 seconds was recorded. If an animal stayed on the beam for the entire 60 seconds, a score of 5 was recorded (1 score was given every 12 seconds). A camera was used to record each trial, and traverse time and falls were used to measure vestibulomotor function (n=8 per group) [20,25].
Cerebrospinal fluid collection from cisterna magna and MMP-9 enzyme-linked immunosorbent assay
A single cerebrospinal fluid (CSF) sample was collected from the animals 72 hours after the induction of TBI (n=8 per group). Briefly, animals were mounted on a stereotaxic device (Stoelting), and local anesthesia was applied (0.25 mL of 1% lidocaine). A 25-gauge scalpel related to a 1.0 mL syringe was placed vertically and centrally into the cisterna magna, and CSF was gently aspirated, resulting in a 50–100 µL sample CSF sample that was centrifuged for 15 minutes at 1,000 ×g, and the supernatant was immediately stored at –80 °C until analysis. Blood-contaminated CSF samples were discarded. Enzyme-linked immunosorbent assay (ELISA) calculate was performed on the collected CSF samples using a rat MMP-9 ELISA kit (MBS722532) from MyBioSource Inc. [26,27].
Histopathology evaluation
For histopathological evaluation, two rats were randomly selected from each group and sacrificed. Brain samples were obtained, fixed in 10% buffered formaldehyde, sectioned (5μm) with an automatic microtome (Leica), and stained with hematoxylin-eosin 24 hours post-TBI. The animals were anesthetized with thiopental (50 mg/kg IP). Pathological changes were evaluated under a light microscope by two pathologists blinded to the animal group and drug used [20,28].
Data analyses
All data are presented as mean±standard error of mean (SEM). Normal distribution of the data was squared using Shapiro-Wilk’s test. In case of normal data distribution, statistical significance was proven using two-way analysis of variance (ANOVA) monitored by Tukey’s post hoc analysis and one-way ANOVA monitored by Newman-Keuls post hoc test. If the data were not normally distributed, a non-parametric Kruskal-Wallis test was used, as surveyed by Dunn’s test for post hoc analysis. The area under the curve (AUC) was calculated using the formula ΔX×([(Y1+Y2)/2]–baseline), where Y1 is the value of the point 1 measurement and Y2 is the second measurement, and ΔX is the amount of time that passed. The baseline was considered to be 0 in all measurements. After calculating AUCs, the SEM and sample size were analyzed using one-way ANOVA in order to find statistical differences among groups. Statistical significance was set at P<0.05. GraphPad Prism version 5.00 for Windows (Graph Pad, www.graphpad.com) was used for the data analysis. The subsequent symbols represent significant values.
RESULTS
The effect of CQ on neurological scores
The effects of CQ treatment on the VCS score after experimental TBI are shown in Table 2. Repeated two-way ANOVA measures were followed by Tukey’s post hoc test. There was no significant difference between the groups before TBI induction. After TBI, all the groups showed a significant decrease (P<0.001). At 24 hours after TBI, VCS scores in the groups that received CQ 3 and 6 mg/kg exhibited a significant increase compared to the saline and TBI groups (P<0.01). At 48 hours after TBI, both groups that received either CQ 3 or 6 mg/kg showed a significant increase in VCS scores compared to the saline or TBI groups (P<0.01). In addition, the group that received CQ 1.5 mg/kg was not significantly different from the saline group at 48 and 72 hours after TBI. Lastly, 72 hours after TBI induction, both groups that received either CQ 3 mg/kg or 6 mg/kg showed a significant increase compared with saline or TBI groups (P<0.05 and P<0.01). On the day of the measurement, a difference between the treated groups was also observed. At 48 and 72 hours after TBI induction, the CQ 6 mg/kg-treated group had significantly higher VCS scores compared to the group that received CQ 3 mg/kg (P<0.01). The AUC analysis of the VCS score yielded similar results (Table 2). All groups were significantly different from the saline group (P<0.01). Groups that were treated with CQ 3 and 6 mg/kg had significantly larger AUC compared to the TBI or saline groups (P<0.05 and P<0.01, respectively).
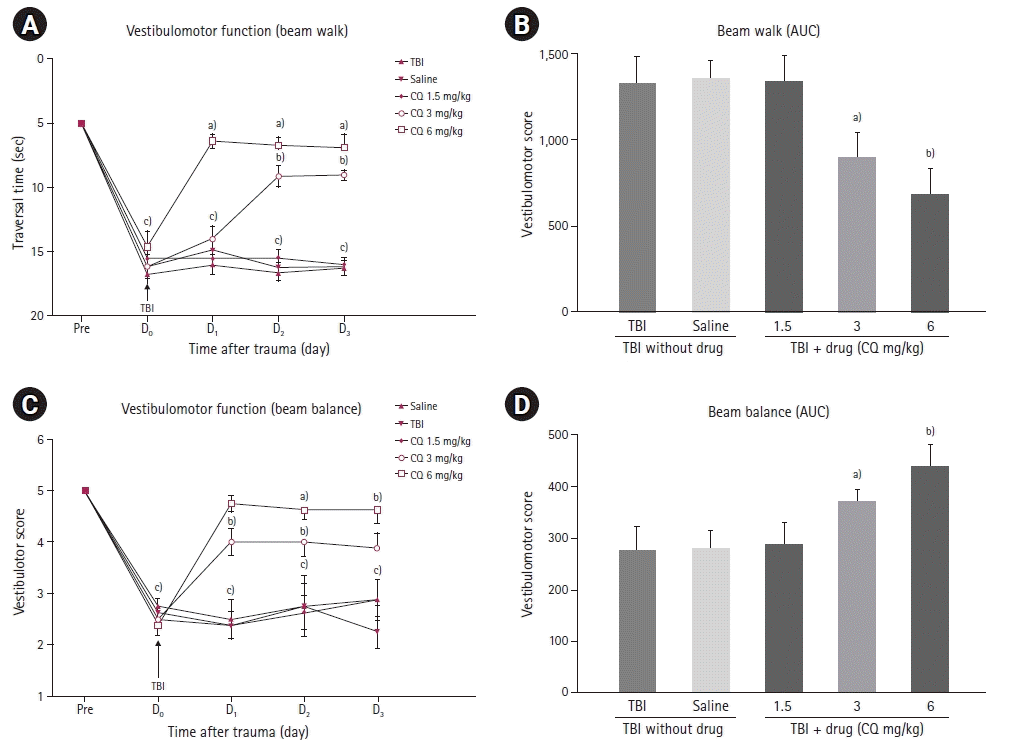
Effect of CQ on beam balance and beam walk tests
The effects of CQ treatment on beam balance and beam walk tests after experimental TBI indicated a repeated measures two-way ANOVA followed by Tukey’s post hoc test. Beam walk and beam balance tasks were used to examine the effect of therapeutic interventions on the vestibulomotor system. Prior to the intervention, all the groups were similar. After TBI, all groups showed significantly lower beam walk scores (Fig. 1A) (P<0.001). This result persisted for 24 hours after TBI. Forty-eight hours after TBI injury, groups treated with either CQ 3 or 6 mg/kg had significantly better beam walk scores than the TBI or saline groups (P<0.05, P<0.01, respectively). This improvement was sustained until the last day of measurement. Similar to the VCS score, 72 hours after TBI injury, the group that received CQ 1.5 mg/kg was not significantly different from saline and TBI groups. The AUC analysis of the data produced similar results (Fig. 1B). The beam balance task yielded similar results to those of the beam walk task (Fig. 1C). Groups that were treated with CQ 3 and 6 mg/kg had significantly better beam balance scores compared to the TBI or saline groups at 48 and 72 hours after TBI injury (P<0.01 and P<0.001, respectively). The observed effect was robust that 48 and 72 hours after TBI injury, the CQ 1.5 mg/kg group was not significantly different from the saline and TBI groups (P>0.05). However, AUC analysis of the data revealed that the CQ 1.5 mg/kg group was not significantly different from the TBI and saline groups (Fig. 1D) (P>0.05).
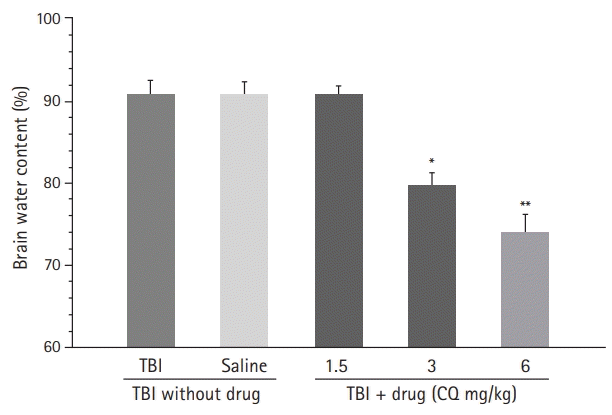
Effect of CQ on brain water content
The effects of CQ treatment on brain water content were repeated using one-way ANOVA followed by the Newman-Keuls post hoc test. The effect of CQ on brain edema, determined by brain water content 24 hours post-TBI, is shown in Fig. 2. The brain water content in the TBI and saline groups was higher (P<0.001). Remarkably, the group that received CQ 1.5 mg/kg was not significantly different from the saline and TBI groups (P>0.05). CQ 6 mg/kg prevented water accumulation in the brain following TBI in comparison with the TBI or saline groups (p<0.01). Although the CQ 3 mg/kg treated group had significantly lower brain water content than the TBI group, the result was not significant compared to the saline and TBI groups (P<0.05).
Effect of CQ on BBB permeability
The effect of CQ on blood-brain permeability by Evans blue dye content of the brain 24 hours post-TBI analysis by repeated measures one-way ANOVA followed by Newman-Keuls post-hoc test is shown in Fig. 3. The Evans blue dye content of the brains in the TBI and saline groups was higher (P<0.001). Remarkably, the group that received CQ 1.5 mg/kg was not significantly different from TBI and saline groups (p>0.05). CQ 6 mg/kg prevented Evans blue dye accumulation in the brain following TBI in comparison with the TBI or saline groups (P<0.01).
Effect of CQ on CSF content of MMP-9
The CSF level of MMP-9 24 hours post-TBI in rats indicated by repeated measures one-way ANOVA followed by Newman-Keuls post hoc test is shown in Fig. 4. An increase in MMP-9 level was observed in the TBI and saline groups (P<0.001). CQ 6 mg/kg was not significantly different between the TBI and saline groups (P>0.05). In addition, CQ 1.5 and 3 mg/kg had significantly lower MMP-9 levels than the TBI and saline groups (P<0.01). There was a significant difference between the CQ1 .5 and 3 mg/kg groups and the saline and TBI groups (P<0.01).
Effect of CQ on histopathological changes
The histopathological alterations in the different groups are shown in Fig. 5. The effects of CQ treatment on histopathological changes were repeated using one-way ANOVA followed by the Newman-Keuls post hoc test. Normal histological architecture of the brain cortex was evident in the saline group. Normal neurons with basophilic, euchromatic, and oval-shaped somas are evident. Most of the neurons in the TBI and saline groups were edematous and irregular in shape with dark nuclei in the TBI and saline groups (Fig. 5A and B). Perivascular edema and necrotic neurons were also observed. Endothelial cell expansion and blood vessel congestion were also observed. No significant histopathological changes were observed in the saline or TBI groups (Fig. 5A and B). A considerable number of pathological changes were observed in the CQ 3 mg/kg group, with reduced numbers of degenerated and edematous neurons (Fig. 5D). In the CQ 6 mg/kg group, most neurons were normal in shape, with euchromatic nuclei and clear nucleoli. Endothelial cells, blood vessels, and astrocytes generally had a normal morphology similar to that of the saline group (Fig. 5E). On the other hand, severe pathological alterations similar to the TBI and saline groups were seen in the CQ 1.5 mg/kg group (Fig. 5C).
DISCUSSION
The goal of this study was to determine the effect of CQ on cerebral edema, BBB permeability, and neurological scores in male rats after brain trauma. According to our findings, among rats with concussions, the group that received 6 mg/kg of CQ had the highest VCS score. The level of consciousness score increased on the third day after concussion, with a VCS score comparable to the sham or intact groups. Qin et al. [11] in 2019 discovered that IP injection of 25 mg/kg CQ in male rats following ischemia-induced brain injury improved neurological scores. In 2015, IP injection of 3 mg/kg CQ in male rats reduced cerebral edema, improved cognitive and motor function, inhibited neuronal autophagy, and decreased interleukin-1 and tumor necrosis factor levels in the hippocampus following brain trauma, which is consistent with our findings [10].
Another finding from our study was that 6 mg/kg CQ was more effective than 3 mg/kg CQ in treating brain edema. The lowest effective dose of CQ was 1.5 mg/kg. Lesiak et al. [29] discovered that after treatment with CQ, the median MMP-9 serum level in patients with systemic lupus erythematosus decreased significantly. CQ treatment declines serum MMP-9 level in systemic lupus erythematosus [30]. It has been found that endosomal maturation inhibitors like CQ block expression of MMP-9 thru toll-like receptor-9 inhibition in murine macrophage RAW 264.7 cells [29]. In addition, the activity and expression of MMP-2 and MMP-9 are attenuated by CQ [31]. This finding is consistent with our previous findings on the effect of CQ on MMP-9 expression. In the histopathological study, 5-μm-thick sections of brains from different experimental groups were prepared and stained with eosin and hematoxylin before being examined under a light microscope. It is likely that the effects of CQ on the MMP may be a major indicator of the loss of BBB integrity. Therefore, we observed behavioral changes with higher doses of CQ. However, for MMP-9, these behavioral changes were observed at lower doses of CQ. The reason may be partly related to the antagonistic and inhibitory effects of this drug on the BBB. Moreover, the dose is toxic and cannot prevent autophagy or apoptosis; thus, the effects of neuroprotection cannot be achieved.
As a result, the histological study found that moderate and high doses of CQ have a positive effect on brain tissue healing, as well as a reduction in cerebral edema, axonal desensitization, and microglial proliferation. Beam balance and beam walk tests were used to assess motor function, and the results showed that 3 days after brain trauma, we gradually observed better results in different groups that received low, medium, and high doses of the drug. The treatment groups had better balance and stayed on the bar for a longer period on the third day of the beam balance test, and the results were similar to those of the sham group.
Dai et al. [32] investigated the effects of CQ on Plasmodium berghei-infected mice in a similar experiment. They used a beam balance test to demonstrate that motor function improved after 10 days of treatment with 20 mg/kg CQ. The amount of Evans blue in the BBB was used to evaluate permeability, which is increased in concussions. The experiments discovered that this index decreased significantly in groups that received CQ doses of 3 and 6 mg/kg, indicating that the CQ had a good neuroprotective effect. The findings of Mielke et al. [33] revealed that for six days, IP administration of CQ (45 mg/kg) had a protective effect on the BBB.
Our findings show that trauma-induced concussion causes cerebral edema and BBB destruction, as well as changes in the animal's neurological and balance scores, and increased MMP-9 levels. On the other hand, CQ at doses of 3 and 6 mg/kg reduced the occurrence of abnormal neurological findings. However, at a dose of 6 mg/kg, these changes were more pronounced, with the exception of a reduction in MMP-9 levels, for which the lowest dose (1.5 mg/kg) had the greatest effect. Contrary to the other results, the high dose of CQ in this case had no effect on this index. Based on our findings and observations, CQ has neuroprotective effects on the brain, and as a result, may help mitigate the effects of brain trauma. Lastly, we propose that the anti-inflammatory effects and CQ neurogenesis are due to the decreased CSF MMP secretion.


 , swollen astrocyte; ★, blood vessel; *, degenerated neuron; →, edematous neuron; ↗, endothelial cell;
, swollen astrocyte; ★, blood vessel; *, degenerated neuron; →, edematous neuron; ↗, endothelial cell;  , Normal neuron.
, Normal neuron.
 XML Download
XML Download