

 PDF
PDF Citation
Citation Print
Print



Introduction
Proteinuria is a well-known risk factor for progressive renal impairment and cardiovascular disease. Kidney damage due to proteinuria is thought to be caused by progressive proliferation due to increased glomerular capillary pressure and plasma protein filtration. Therefore, if persistent proteinuria is detected, kidney biopsy is considered for the diagnosis and treatment of the underlying disease. However, for diseases in which renal damage is not caused despite the patient showing symptoms of proteinuria, there may be cases in which various treatments used for protection of renal damage are unnecessary. Recently, with the development of next generation sequencing (NGS) technology, a mutation in the cubulin (CUBN) gene was detected in patients with hereditary kidney diseases that have chronic proteinuria. Although this disease leads to proteinuria, there are many questions about its progression to renal impairment. In this review, we focus on cubilin and the latest views on the prognosis of the CUBN mutation.
Pathophysiology of proteinuria
Proteinuria is mainly divided into glomerular proteinuria and tubular proteinuria based on the origin of proteinuria. Dysfunction of the glomerular filtration barrier results in glomerular proteinuria, mainly albumin, which has a molecular weight of approximately 67 kDa. In contrast, tubular proteinuria is caused by the dysfunction of tubular reabsorption. It is composed of low molecular weight proteins, mainly β2 microglobulin and α1 microglobulin. Furthermore, albumin accounts for less than half of tubular proteinuria.
The passage of molecules through the glomerular basement membrane (GBM) is size- and charge-dependent. The GBM has a pore size of 4 nm and a negative charge due to the presence of glycoaminoglycans. Therefore, negatively charged and large molecules cannot pass through the GBM easily. Albumin has a negative charge and has an ellipsoid shape with a 14-nm width and 3.8-nm length. Because the length (3.8 nm) of albumin is shorter than the GBM pore size (4 nm), small amounts (22–32 mg/L) of albumin can be filtered through the GBM. The albumin is then reabsorbed at the early and late parts of the proximal tubules and descending part of the straight tubules. Finally, only approximately 100 mg of albumin is secreted per day [1,2].
Genetic aspects of proximal tubular reabsorption
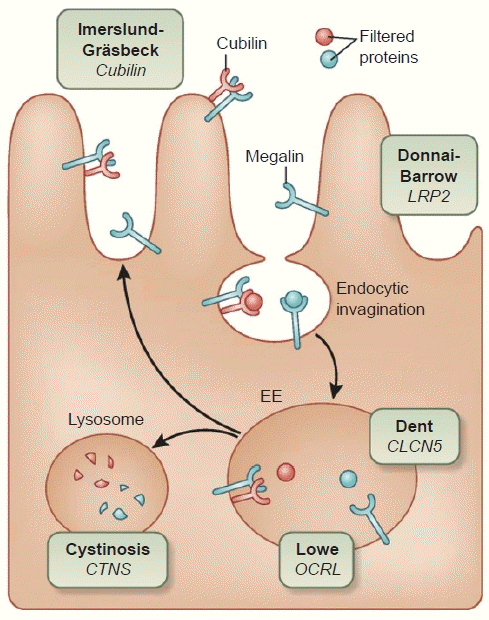
Proximal tubules have receptors called megalin and cubilin-amnionless complex (CUBAM) at the brush border. These receptors bind to several ligands, including albumin, and transfer these ligands into the endosome in the proximal tubular cytoplasm via clathrin-mediated endocytosis. Subsequently, these ligands are degraded by the lysosome or undergo transcytosis via the neonatal Fc receptor [3-5].
Several genetic tubular diseases develop via the mutations in this process. Donnai-Barrow syndrome (OMIM #222448) is caused by a mutation in megalin (gene name: LRP2), Imerslund-Gräsbeck syndrome or megaloblastic anemia 1 (IGS or MGA1) (OMIM #261100) by a mutation in cubilin (gene name: CUBN), Dent disease (OMIM #300009) by a mutation in endosomal chloride voltage-gated channel 5 (gene name: CLCN5), Lowe syndrome (OMIM #3090000) by a mutation in endosomal inositol polyphosphate-5-phosphatase (gene name: OCRL), and nephrotic cystinosis (OMIM #219800) by a mutation in lysosomal cystinosin (gene name: CTNS) (Fig. 1) [6-11].
Molecular structures of megalin, cubilin, and amnionless
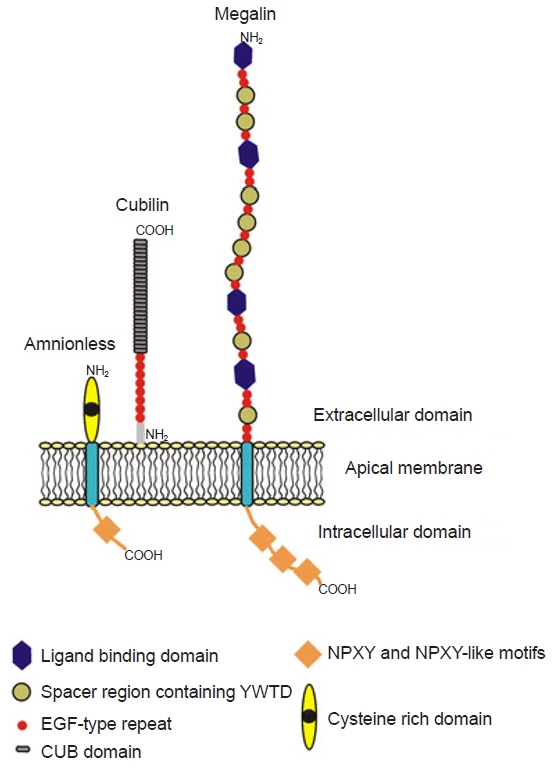
Cubilin, an albumin receptor, was first described by Birn et al. in 2000 [12]. It is an extracellular glycoprotein approximately 460 kDa in size, and the gene encoding cubilin is located on chromosome 10p12.33-p13 in humans. Cubilin has 110 amino acid N-terminals, 8 epidermal growth factor-like domains, and 27 complement C1r/C1s, Uegf, and Bmp1 (CUB) domains (Fig. 2) [1]. Because cubilin does not have a transmembrane domain, not fully understood, it interacts with amnionless (CUBAM) for endocytosis.
Clinical manifestations of CUBN mutation
Cubilin is expressed in podocytes, proximal tubules, and the small intestine. As described above, cubilin plays a role in albumin reabsorption in the proximal tubule. Furthermore, it is a receptor of the intrinsic factor-vitamin B12 (IF-B12) complex in the small intestine. Therefore, patients with CUBN mutations can develop proteinuria and/or MGA.
IGS (or MGA1), caused by mutations in CUBN, is a rare, autosomal recessive disorder that is characterized by selective intestinal vitamin B12 malabsorption and results in MGA. It also has low molecular weight proteinuria. In most cases, proteinuria in IGS is sub-nephrotic range proteinuria and is characterized by more than 50% albuminuria [9,10].
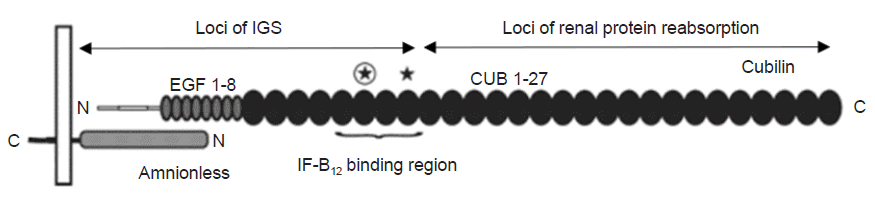
Fig. 3 shows the currently known loci of the CUBN mutation. Most mutation sites of CUBN in IGS are located at the N-terminal of cubilin and affect the interaction sites with amnionless or the IF-B12 binding CUB domains 5–8. However, in patients with only proteinuria, almost all variants were located after the IF-B12 binding domain. This suggests that C-terminal CUB domains are crucial for renal protein reabsorption [10].
The history of studies on CUBN mutations
Since 1999, IGS with MGA and proteinuria has been reported [13-15]. In 2011, Ovunc et al. [16] reported a homozygous frameshift mutation in CUBN in two siblings from consanguineous parents with intermittent nephrotic-range proteinuria by exome sequencing. Furthermore, a meta-analysis through a genome wide association study (GWAS) in the general and diabetic population highlighted the relationship between CUBN single nucleotide polymorphism and proteinuria in the same year [17].
Since 2015, large-scale cohort studies for genetic diagnosis of patients with proteinuria have shown a certain number of patients with CUBN mutations [18-23]. One of these was the result of three large cohort studies on CUBN mutations by Bedin et al. in 2020 [21]. A total of 2,216 patients were enrolled in three cohort studies. In the first cohort (genetic kidney disease cohort I), NGS was performed on 759 patients whose suspected genetic causes of renal disease included steroid resistant nephrotic syndrome (SRNS), Alport syndrome (AS), nephronophthisis, congenital anomalies of kidney and urinary tract, tubulointerstitial nephritis, polycystic kidney disease, and renal tubular disorder. For the second cohort (genetic kidney disease cohort II), NGS was performed on 1,350 patients whose suspected genetic cause of proteinuria included SRNS and AS, and for the third cohort (chronic PU cohort), NGS was performed on 107 patients with chronic proteinuria. In these three cohort studies, biallelic CUBN mutations were observed in 39 patients. The 39 patients with biallelic CUBN mutations received a diagnosis between 0 and 36.6 years and did not show MGA or renal impairment during the follow-up period (age at last follow-up: 0–71.7 years). Their proteinuria was less than 1 g/day, and the albumin content in the urine protein was more than half. Unlike other tubular proteinuria including caused by LRP2 mutation, β2 microglobulin was almost absent or low. Kidney biopsy was conducted on a total of 19 patients. One patient in the second cohort showed early focal segmental glomerulonephritis (FSGS), and the remaining showed no specific finding or minimal lesions. All patients with the CUBN mutations in the third cohort showed no protein lowering effects when treated with angiotensin converting enzyme inhibitor (ACEi). Furthermore, almost all variants of the CUBN gene were located after the IF-B12 binding domain.
In this papaer, another large-scale meta-analysis of existing GWAS was conducted with the general population. In previous studies, four C-terminal variants (p.A1690V, p.N2157D, p.A2914V, and p.I2984V) were shown to have strong associations with albuminuria using GWAS. According to structural modeling, these four GWAS variants have the potential to disturb the CUB domain stability or ligand binding. Two more meta-analyses were conducted: a large meta-analysis of the CKDGen Consortium’s population-based cohorts consisting of 331,340 to 597,710 individuals, and a smaller one on an independent cohort consisting of 13,550 individuals with and without type 2 diabetes mellitus (T2DM). The results showed that the population with four C-terminal variants had a tendency of exhibiting high glomerular filtration rate (GFR), despite having albuminuria. Thus, this paper mentioned that the C-terminal CUBN mutation may have a renal protective effect.
In addition, in a protein-coding genetic variation analysis for 60,000 people in 2016 [24], CUBN and AMN (which encodes amnionless) showed very low loss-of-function intolerance scores. These data strongly indicate the benign nature of albuminuria associated with C-terminal CUBN variants.
The second large-scale study included a cohort of a Spanish group in 2021 [22]. Genetic testing was performed on 347 families with persistent proteinuria with a suspected monogenic cause. Among them, CUBN biallelic mutations were observed in 15 patients in 12 families. The age at diagnosis of proteinuria ranged between 9 months and 44 years. Furthermore, 54% of urine protein was albumin. The follow-up period was 7 years on average, and no improvement in proteinuria was observed after treatment with ACEi. Among 15 biopsies, 13 were normal, but two patients had more severe phenotypes. One patient was diagnosed as SRNS with minimal change disease through the kidney biopsy. This patient used prednisolone and cyclophosphamide and showed normal kidney function and blood pressure during follow-up, but proteinuria persisted. Another patient was diagnosed as FSGS on the biopsy and showed hypertension, hyperuricemia, decreased GFR, hypertension induced cardiomyopathy, T2DM, and obesity during the follow-up period. The authors thought that the progress of these patients was due to comorbidities.
Meanwhile, Yang et al. in China [25] described three FSGS patients with CUBN mutations in 2022. Patients were diagnosed at the age of 6 years, 8 years, and 11 years and showed no MGA. They had mild proteinuria, and their urine protein to creatinine ratio were 0.3–0.5, β2 microglobulin was normal or slightly high, and urine protein electrophoresis showed more than 50% albumin. The serum albumin level of the patients was normal and showed 1–2 segmental sclerosis in kidney biopsy. Two patients were accompanied by interstitial fibrosis, and a crescent was also observed in these two cases. All electron microscopy showed foot process effacements. All CUBN mutations were biallelic, and all were located close to the C-terminal. These three patients were administered tacrolimus after FSGS was confirmed in the biopsy, and their protein to creatinine ratios decreased slightly during the follow-up period of more than 3 months. The authors suggested that albuminuria probably originated not only from proximal tubular malabsorption but also from podocyte dysfunction in patients with CUBN mutations. This is because patients showed FSGS with pathological changes in podocytes and reduced proteinuria after administration of tacrolimus. In addition, one of four patients in the genetic kidney disease cohort II with CUBN mutations in the study of Bedin et al. also showed FSGS [21]. Furthermore, some studies showed that cubilin is expressed in rat and human podocytes and mediates albumin endocytosis in human podocytes. Another line of evidence suggests that cubilin has a higher binding affinity for albumin compared to that for megalin. Megalin can function as a sensor of albumin to affect cell survival via the phosphoinositide 3-kinase/protein kinase B pathway. If a large quantity of albumin binds to megalin due to cubilin dysfunction, it can promote cell apoptosis [28-30]. Furthermore, patients with FSGS had at least one relatively serious mutation in one allele. These include nonsense mutations, insertions, deletions, or mutations in splice sites which led to frameshift mutations or protein truncation.
The most recent report on CUBN mutations was a cohort study by a group in Turkey published in 2022 [23]. Through genetic tests performed on 158 patients with SRNS or chronic proteinuria, the authors found a total of six CUBN mutations, all of which were C-terminal. Kidney biopsy was performed in four of six patients, one of whom was diagnosed with FSGS, and a homozygous nonsense mutation was observed in this patient. ACEi or angiotensin II receptor blocker (ARB) had no therapeutic effects on all patients, and FSGS patients were also unresponsive to prednisolone, cyclosporine, and rituximab treatment. All patients showed normal renal function during follow-up even after all treatments were stopped.
Is proteinuria caused by CUBN mutation benign?
Table 1 shows a summary of studies on CUBN mutations. There is a limitation that the total number of patients with CUBN mutations and the follow-up duration was not sufficient to conclude that the CUBN mutation is benign and does not progress to chronic kidney disease (CKD) based on these studies alone. However, no patients progressed to CKD except for one patient with renal impairment due to comorbidity in these studies [22]. In addition, some reports suggested the hypothesis that the CUBN mutation could be benign [21,24]. The hypothesis about the reno-protective effect of CUBN mutation like sodium glucose cotransporter 2 inhibitor provided hope that the CUBN mutation could be benign [31]. However, patients exhibit FSGS on histological examination (Table 1) [18,21,23-25] and cubilin is expressed not only in renal tubules, but also in podocytes [28,29]; thus, concluding that CUBN mutation is benign is a challenging task. Future studies will have to be conducted with long-term follow-up of more patients for more concrete evidence.
When do we suspect CUBN mutation and what genetic tests should we run?
In patients with asymptomatic proteinuria who do not have hypertension, hypoalbuminemia, if the proteinuria is in the sub-nephrotic range, β2 microglobulin is low, and albuminuria is greater than 50%, CUBN mutation may be suspected. If CUBN mutation is suspected, genetic kidney disease screening for CUBN mutation by whole exome sequencing or a kidney disease targeted gene panel test that includes CUBN is recommended prior to kidney biopsy.
Conclusion
Proteinuria, due to CUBN mutation, is currently presumed to be caused by kidney tubular dysfunction, which leads to chronic sub-nephrotic proteinuria with high urine albumin ratios. Therefore, if a patient with chronic sub-nephrotic proteinuria had high urine protein to creatinine ratios and normal kidney function, genetic analysis for the CUBN mutation must be considered. To date, there is no clear verification that patients with the CUBN mutation do not need ACEi or ARB for kidney protection because it is unclear if it is a benign disease. However, if a CUBN mutation is diagnosed, at least kidney biopsy and the use of immunosuppressants can be avoided. Future studies must be conducted to conclude whether this disease shows a benign course through long-term follow-up.
 XML Download
XML Download