

 PDF
PDF Citation
Citation Print
Print



Introduction
Nephron, a microscopic structural and functional unit of kidney, is composed of glomerulus, vasculature, tubules, and interstitium. Renal tubules are responsible for homeostasis of body fluid, electrolyte, and acid-base conditions [1,2]. The tubules modulate reabsorption and secretion of fluid and solutes by passive or active mechanisms [1-3].
Renal tubules consist of four main segments with different anatomies and functions: proximal convoluted tubule (PCT), thick ascending limb of Henle’s loop (TAL), distal convoluted tubule (DCT), and collecting duct (CD). Each segments perform distinct functions with specific transporters. Dysfunctions of these tubular transports are called tubulopathies [1-3].
Tubulopathies have either inherited or acquired etiologies [3,4]. This review focused on inherited tubulopathies that generally manifest in pediatric period. Mutant variants encoding transporters in any tubular segments can cause inherited tubulopathies and determine their distinguishing clinical features [2]. Remarkable advances in genetic diagnosis have expanded our knowledge of inherited tubulopathies.
This review tries to present clinical clues to approach and diagnose inherited tubulopathies. Simplification of categorizing the tubular diseases could allow clear diagnosis and prompt management. Moreover, present review clarifies the recently defined concept of autosomal dominant tubulointerstitial disease (ADTKD) and introduces a few novel therapies for tubulopathies updated lately.
Clinical and genetic approach to tubulopathies
Inherited tubulopathies are commonly discussed in academic sessions with details of a particular disease. However, there are numerous diseases associated with various transporters and tubular segments and the incidence of tubular diseases is relatively low comparing with the other glomerular diseases. Due to the diversity and rarity, diagnosing the inherited tubulopathies remains challenging.
In the practical field, distinct clinical presentations are substantial for diagnosing tubulopathies. Nephrologists should focus on manifestations identifying affected segments and transporters other than the common features of polyuria, polydipsia, and growth failure. Part I and II describe functions of different tubular segments and categorize tubular diseases according to the key features.
Part I. Principal functions of the tubular segments
Table 1 describes the functions of each tubular segment. PCT plays a major role in reabsorbing most of water and solutes. These solutes include glucose, bicarbonate, amino acids, low-molecular-weight proteins, calcium, phosphate, and citrate. Na+-K+-ATPase, the main transporter of PCT, has high energy requirement. Thus, PCT segment is vulnerable to hypoxic conditions and impairment of energy supply such as mitochondrial cytopathies and inborn error of metabolism [2-5].
TAL and DCT can regulate urinary concentration by reabsorbing sodium and chloride via Na+-K+-2Cl– cotransporter (NKCC2) and Na+-Cl– cotransporter (NCC), respectively. These two segments also contribute to calcium and magnesium homeostasis via paracellular and transcellular mechanisms [2,3,5].
CD consists of two cell types: principal cells and α-intercalated cells. In principal cells, the epithelial sodium channel (ENaC) modulated by aldosterone, can reabsorb Na+ and secrete K+ while arginine vasopressin receptor 2 (AVPR2) and aquaporin 2 (AQP2), controlled by antidiuretic hormone (ADH) can reabsorb free water. Acid or bicarbonate is excreted through α-intercalated cells [2,3,5].
Part II. Clinical approach to inherited tubulopathies
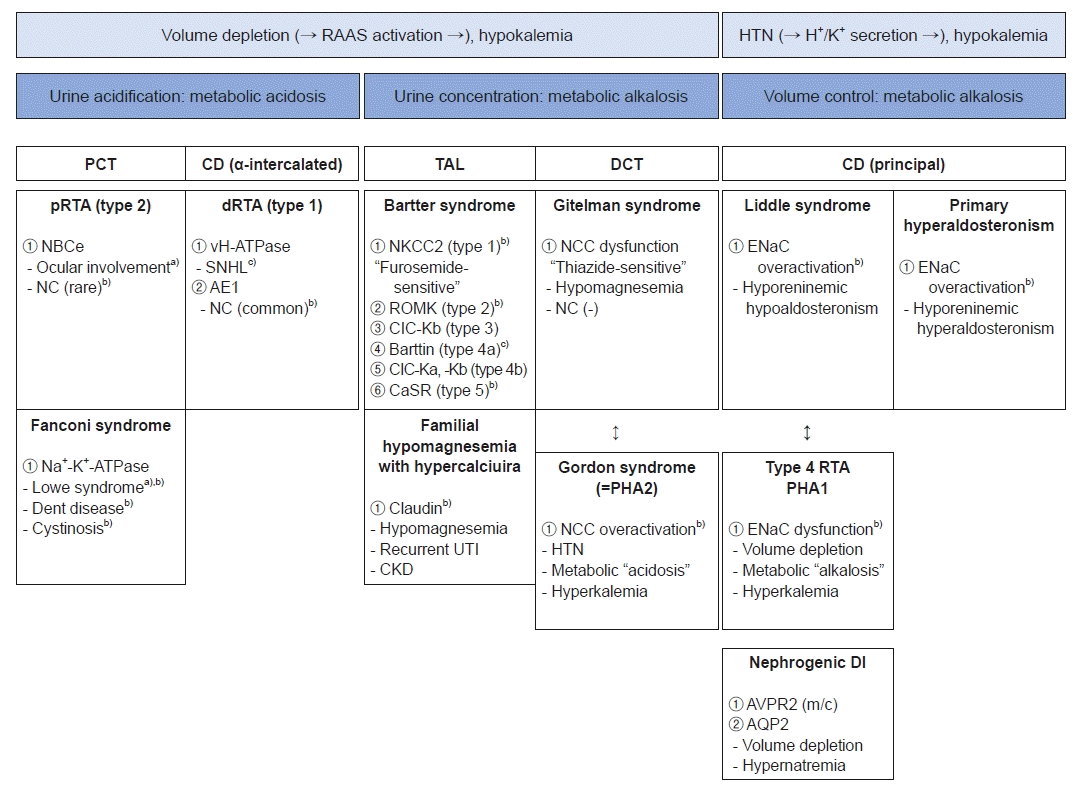
When evaluating patients clinically suspicious of underlying inherited tubulopathies, it is important to differentiate clinical conditions with simple categorizations before localizing the affected tubular segment. Patients with inherited tubulopathies can be classified according to (1) the volume of body fluid and (2) acid-base status. Fig. 1 describes early differential diagnosis based on volume and acid-base status sequentially. Although some patients show hypertension, most with tubulopathy show volume depletion due to the malabsorption. Activation of renin-angiotensin-aldosterone system (RAAS) then causes hypokalemia. Metabolic acidosis is accompanied by defect of urine acidification in which PCT and α-intercalated cells of CD play a major role. Meanwhile, abnormal urine concentration can lead to metabolic alkalosis in TAL and DCT, target segments of diuretics [3].
Hypertension presents in a few types of tubulopathies characterized by mineralocorticoid excess or activation of aldosterone-sensitive ENaC in the CD. Sodium and water retention can result in excretion of potassium and hydrogen ion [3,6].
Proximal convoluted tubule
Proximal renal tubular acidosis (pRTA, type 2 RTA)
Impaired function of sodium-bicarbonate cotransporter caused by SLC4A4 variants wastes bicarbonate, leading to metabolic acidosis. Unlike distal renal tubular acidosis (dRTA), nephrocalcinosis can be prevented by citraturia, one of the protective factors against urinary stone [3,7]. Patients with pRTA need ophthalmologic examination since ocular involvements such as cataract and glaucoma are frequent [3,8].
Fanconi syndrome
Na+-K+-ATPase dysfunction is related to wasting of all solutes reabsorbed in PCT. This mechanism results in glycosuria, proteinuria, hypercalciuria with nephrocalcinosis, hypophosphatemia along with rickets, and growth retardation [9]. Lowe syndrome (OCRL mutation) and Dent disease (type 1 by CLCN5 variants and type 2 by OCRL mutation) are typical disorders manifesting Fanconi syndrome. Both diseases present proteinuria, marked hypercalciuria with nephrocalcinosis and progressive chronic kidney disease (CKD). Lowe syndrome is highly associated with early onset of extra-renal manifestations including ocular (cataract, glaucoma, and visual disturbance) and neurological involvements (hypotonia, seizures, intellectual disabilities, and behavioral abnormalities) [10]. Dent disease type 2 is also caused by OCRL mutation. However, it is less likely to be accompanied by extra-renal symptoms. Thus, it is described as a “mild Lowe syndrome” [10,11]. Cystinosis and galactosemia can also present with Fanconi syndrome due to accumulation of toxic metabolites [9].
Thick ascending limb of Henle's loop
Bartter syndrome
Bartter syndrome (BS) has five subtypes with discrete transporters affected. Type 1 BS is associated with furosemide-sensitive NKCC2 (SLC12A1 mutation) on the apical membrane. Accordingly, chronic exposure to furosemide can bring out Bartter-like symptoms. Type 2 BS is associated with renal outer medullary potassium (ROMK) channel (KCNJ1 mutation), another transporter on the apical membrane. Nephrocalcinosis is common in type 1 BS and type 2 BS as the disrupted channels on the apical membrane of TAL can affect paracellular mechanisms of calcium reabsorption by generating electrochemical gradient [12]. BS types 3, 4a and 4b are due to dysfunctions of chloride channel (CIC)-Kb (CLCNKB), Barttin subunit (BSND), and CIC-Ka (CLCNKA), respectively. Type 4a BS is associated with sensorineural hearing loss (SNHL) as the Barttin subunit copresent in the inner ear [5]. Type 5 BS (MAGED2) is associated with mutations of calcium sensing receptor. It is directly related to nephrocalcinosis. The onset of BS ranges from antenatal to early childhood depending on the subtype. Usually manifesting in antenatal or neonatal period, type 3 BS shows the latest onset in childhood. Interestingly, type 5 BS presents transiently and resolves within the first 3 months of life [13,14].
Familial hypomagnesemia with hypercalciuria
Additionally, on the tight junction, protein called claudin can modulate paracellular calcium and magnesium reabsorption. Mutations in claudin can cause familial hypomagnesemia with hypercalciuria. Patients with this syndrome have higher risk of recurrent urinary tract infection and progressive CKD [15].
Distal convoluted tubule
Gitelman syndrome
Gitelman syndrome (GS) is caused by mutations in SLC12A3 gene encoding NCC. NCC is a thiazide-sensitive transporter. Thus, GS exhibits metabolic alkalosis with hypokalemia, similar to BS. However, unlike BS, GS has relatively late onset in childhood or adolescence and presents hypocalciuria and symptomatic hypomagnesemia [13,14]. GS is due to under-expressed magnesium channels of TRPM6, triggering calcium resorption alternatively [12].
Gordon syndrome (also known as pseudohypoaldosteronism type 2)
The mirror image of GS is Gordon syndrome, also known as pseudohypoaldosteronism type 2 (PHA2). This disease is caused by overactivation of NCC with WNK4, WNK1, CUL3, and KLHL3 mutations. Hypertension results from increased sodium and chloride reabsorption, subsequently leading to volume expansion. Compensatory suppression of sodium reabsorption in CD can reduce potassium and hydrogen secretion which can manifest as metabolic acidosis with hyperkalemia [3,8].
Alpha-intercalated cells of CD
Distal renal tubular acidosis (dRTA, type 1 RTA)
Dysfunctions of vacuolar type (H+)-ATPase (vH-ATPase; ATP6V1B1, QTP6V0A4, and FOXI1 mutations) and anion exchanger 1 (SLC4A1 and WDR72 mutations) for excreting hydrogen ion can cause metabolic acidosis. The acidic status and alkalic urine can contribute to bone resorption, hypercalciuria and nephrocalcinosis. Since the vH-ATPase copresent in inner ear, patients with disrupted vH-ATPase are vulnerable to SNHL [3,5].
Principal cells of CD
Primary hyperaldosteronism
Various conditions including genetic diseases and adrenal hyperplasia are associated with hyperaldosteronism and the following suppression of renin [3]. Familial hyperaldosteronism is one of the genetic conditions presenting hypertension with high aldosterone level [6]. There are four types of familial hyperaldosteronism and the causative genes are CYP11B1, CLCN2, KCNJ5, and CACNA1H, corresponding sequentially for types 1-4 [6].
Liddle syndrome
Variants of SCNN1B/G, a causative gene of Liddle syndrome, can inhibit the degradation of ENaC for maintaining increased activity of the channel. Since the mechanism is independent of RAAS, both renin and aldosterone are suppressed [3,6]. Excess sodium causes hypertension, extrusion of potassium via Na+-K+-ATPase, and metabolic alkalosis by secretion of hydrogen ion through ROMK channel and vH-ATPase [6].
Type 4 RTA and pseudohypoaldosteronism type 1 (PHA1)
Opposite to Liddle syndrome, impaired sodium reabsorption of ENaC and reduced excretion of hydrogen and potassium can occur by aldosterone deficiency or resistance. The former condition of aldosterone deficiency is referred to as type 4 RTA. Urosepsis or obstructive uropathy is a common cause of type 4 RTA in children. Spironolactone use and congenital adrenal hyperplasia can also be causes of type 4 RTA [8]. Genetic forms of type 4 RTA are known as PHA1. However, these patients typically present with hyper-reninemic hyperaldosteronism since PHA1 is due to mineralocorticoid resistance [8].
Nephrogenic diabetes insipidus
Loss-of-function variation of AVPR2 is the most common cause of nephrogenic diabetes insipidus (NDI), followed by AQP2 variants. Patients with NDI are resistant to ADH, leading to volume depletion and hypernatremia (serum osmolality >urine osmolality) [3,16]. Most of affected patients present with failure to thrive during the first few months of life [16]. Absence of antenatal history of polyhydramnios helps to distinguish NDI from other polyuric disorders, such as BS [16].
Part III. ADTKD and whole exome sequencing as a diagnostic tool
Unexplained familial clustering, which implicates the underlying genetic kidney disease, has been detected among CKD patients. With increasing knowledge about inherited kidney disorders, monogenic causes have been identified in a few patients. Although the etiology is still not established for most cases, monogenic disorders were diagnosed in about 10% of CKD patients [17,18]. Autosomal dominant polycystic kidney disease (ADPKD) is the most common genetic kidney disease, followed by type IV collagen disorder [17,18]. ADTKD, a recently defined disease entity, has also been suggested as one of the common monogenic kidney diseases [19].
Different from ADPKD and type IV collagen disorder, some patients show characteristic features of tubulointerstitium-limited involvements with definite familial histories and gradual progression to end-stage kidney disease. From these nonglomerular autosomal dominant kidney diseases, genetic mutations of UMOD, REN, HNF1β, and MUC1 have been detected [17,18]. Additional identifications are in progress.
Various terms have been used over the years to describe this disease group. However, old terms including medullary cystic kidney disease and familial juvenile hyperuricemic nephropathy did not point out nor correspond to overall characteristics of the disease. Establishing uniform diagnostic criteria and nomenclature was unavoidable. Therefore, in 2015, Kidney Disease Improving Global Outcomes standardized the term as ADTKD. According to the affected gene, the ADTKD is specified into ADTKD-UMOD, -MUC1, -REN, -HNF1β, and -NOS (not otherwise specified) [17].
Clinical findings of ADTKD correspond to the following diagnostic criteria: (1) autosomal dominant inheritance; (2) progressive loss of kidney function; and (3) bland urinary sediment (normal or mild microscopic hematuria). Glomerular function remains intact so that proteinuria should be absent or mild. Renal histology reveals degenerative changes involving only tubules and interstitium. Sonographic findings including small-sized kidney and renal cysts are not pathognomonic signs. Confirmative diagnosis is made with compatible familial history of CKD and fulfilling clinical characteristics or with demonstration of a mutation for one of the four known genes [17].
Table 2 describes the specific features of each subtype of ADTKD [19]. ADTKD-UMOD is the most common subtype. Accumulation of abnormal uromodulin in TAL can inhibit NKCC2 resulting in decreased reabsorption of sodium. Compensatory increase in reabsorption of PCT can reduce urinary secretion of uric acid and cause the typical symptoms of hyperuricemia and early gout. ADTKD-MUC1 also deposits abnormal mucin 1 in TAL. However, the pathophysiology is incompletely known. Unique features have not been noted except gout. For ADTKD-HNF1β, renal manifestations may present early in childhood as congenital anomalies of kidney and urinary tract. Since hepatocyte nuclear factor 1β is involved in fetal development of multi-organs, patients might have numerous extra-renal manifestations such as genital abnormalities, abnormal liver function, diabetes mellitus, and autism spectrum disorder. Interestingly, de novo mutation is estimated to account for up to 50% cases of ADTKD-HNF1β. Incomplete penetrance is also common. These are the factors interrupting the diagnosis of ADTKD-HNF1β with confusing pedigree. Hyporeninemic hypoaldosteronism of ADTKD-REN can result in childhood anemia, hypotension, and hyperkalemia.
Genetic diagnosis is generally recommended for certain diseases. It can benefit diagnosis considering cost and ethical issue. Genetic analysis is currently the only way to definitively prove ADTKD, although disease-specific therapies are currently unavailable [17]. Still, children with subtypes of REN and HNF1β might benefit from early conservative management of anemia, hypotension, and other extra-renal symptoms, although UMOD and MUC1 related ADTKD have no distinct symptoms requiring interventions except gout [20]. Of diverse types of genetic testing, whole exome sequencing (WES) is an adequate diagnostic tool for ADTKD due to the incomplete recognition of the disease entity. It is impractical to clinically differentiate the subtypes of ADTKD so far. Moreover, only 50% to 60% of suspected ADTKD patients show the discovered mutations of four genes, indicating that a negative genetic testing result does not exclude the diagnosis [17,18]. Therefore, applying WES for diagnosis ADTKD would be more appropriate than sequencing gene panels.
Sinha et al. [21] performed research on the utility of WES for confirming other inherited tubular disorders in addition to ADTKD. WES was performed for 77 pediatric patients who were clinically diagnosed renal tubular diseases at median age of 48 months. Overall diagnostic yield was 70%. Revision of clinical diagnosis was made in 18%, higher comparing a rate of 4% in previous research of Ashton et al. by Sanger sequencing [22]. WES also directly impacted the clinical management for 31% of the cohort and helped reveal other associated pathologies such as SNHL [21].
Apparently, WES performs efficient verifications of inherited tubulopathies especially ADTKD. However, gene panels still could be a good option for patients with distinctive and typical manifestations of particular tubulopathies. Further discussion is needed to suggest more suitable types of genetic testing for inherited tubulopathies considering the cost and ethical issues such as undesirable reveal of private information including secondary findings or familial histories.
Part IV. Gene therapy in tubulopathies
Multiple genes have been implicated as causes of genetic kidney disease including tubulopathies recently. The necessity of disease-specific therapy targeting the causative gene is emerging along with the acceleration of genetic diagnosis in CKD patients [23]. Undoubtedly, the field of gene therapy has markedly improved recently. For kidney diseases, however, relatively little progress has been made toward gene therapy due to several reasons: (1) given the complex structure and over 20 different cell types, it is difficult to optimally target the affected cell type; (2) causative genes of most prevalent genetic kidney diseases are larger than the capacity of adeno-associated vector (AAV) which is the most common vector used for in vivo strategies; and (3) with the most favored intravenous route, most vectors get lost in the liver and 26 nm-sized AAV can hardly pass through the 10 nm-sized slit diaphragm of glomerulus [24].
Consistent and vigorous attempts are being continued to overcome the points that are currently limiting gene therapy in kidney diseases. As the size of genes packed in AAV and an efficient delivery are the crucial points to modify, studies focusing on utilizing smaller genes essential for modifying target genes and alternative delivery strategies such as subcapsular direct injection or retro-ureteral route are in progress [18,24,25].
Despite the difficulties mentioned above, novel therapies for inherited tubulopathies are actively investigated. Here we introduce on-going research on cystinosis and Lowe syndrome.
Cystinosis is an autosomal recessive lysosomal storage disease caused by CTNS variants. Affected patients are characterized by lysosomal accumulation of cystine crystal in multiple organs including kidney, eye, pancreas, and muscles. Current conservative treatment is the use of drug cysteamine which is expensive, inconvenient to take, and not for cure. Cherqui et al. [26,27] have tried ex vivo cell therapy using hematopoietic stem and progenitor cells (HSPC). In phase I/II clinical trial starting from July 2019, autologous HSPCs were gene-modified ex vivo with CTNS-loaded lentivirus vectors and transplanted to patients. The research team of Cherqui [27] provided a recent update in September 2022. Their results revealed significant longitudinal decrease in leukocyte cystine level and corneal cystine crystals in patients treated with the cell therapy. Lowe syndrome, another proximal tubulopathy, is a X-linked multisystemic disorder caused by OCRL gene mutation. Deposits of phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2) can produce excessive actin which blocks endosomal trafficking and result in congenital cataract, intellectual disability, and proximal renal tubular dysfunction [10]. Berquez et al. [28] have focused on rebalancing phosphoinositide signals by phosphoinositide 3-kinase (PI3K) inhibitor. The team performed mouse model research of alpelisib which was conventionally used to treat breast cancer but lately known as PI3K inhibitor. When administered to mice model of Lowe syndrome, alpelisib alleviated actin organizational defect and improved kidney function. Likewise, numerous studies are actively in progress to establish novel therapies not only for the proximal tubular disease but also for other inherited tubular diseases including ADTKD.
Conclusion
Distinct transporters exist in each of four tubular segments. In patients with inherited tubular disorders, phenotypes are determined by affected transporters. Interpreting the status of volume and acid-base will help physicians localize the affected segment. ADTKD, a recently defined disease entity, is one of more common genetic kidney disease. Since much about ADTKD have remained unknown, WES can be a suitable tool for discovering both known and unknown genetic information. Along with advances in genetic diagnosis, disease-specific novel therapies for tubulopathies are currently under development.
 XML Download
XML Download