

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Cancer immunotherapy uses complementary innate and adaptive immune responses to enhance the host’s systemic and selective immunity against tumor cells. In innate immunity, natural killer (NK) cells and myeloid cells recognize and eliminate tumor cells in a major histocompatibility complex (MHC)-independent manner. Whereas innate immunity occurs immediately but does not have antigen specificity, adaptive immunity is antigen-specific and is initiated when antigen-presenting cells (APCs) such as dendritic cells (DCs) present tumor antigens. Naïve T cells recognize tumor-derived antigen epitopes as MHC-peptide complexes presented by APCs and then combine them with T cell receptors (TCRs) to become effector T cells that have cytotoxicity to tumor cells expressing the same antigen.
T cells are the major force of the adaptive immune response, and their potency influences the efficacy of cancer immunotherapy. Disappointing results from recent immunotherapeutic clinical trials for peptide and DC vaccines to induce the endogenous activation of T cells against glioblastomas [21,27,112,227,228,243] suggest that the antitumor immune response induced by those strategies might be insufficient to control tumors. Those trials might also have failed to expand the population of tumor antigen-specific T cells reproducibly and effectively. The tumor-associated antigens (TAAs) used in the vaccines can be somewhat expressed in normal tissues, so the immune system might recognize them as self-antigens, which would decrease the T cell response through the mechanisms of immune tolerance, which remove T cells with high affinity to self-antigens [31,196]. T cell exhaustion could be another reason for the disappointing results. T cell exhaustion is a state of T cell dysfunction in environments such as chronic infections and cancer that involve chronic antigen exposure and lack of appropriate assistance from helper T cells [247]. Exhausted T cells increase their expression of inhibitory receptors, including programmed death-1 (PD-1), cytotoxic T lymphocyte antigen-4 (CTLA-4), lymphocyte activation gene-3 (LAG-3), and T cell immunoglobulin domain and mucin domain protein-3 (TIM-3), and they decrease their production of effector cytokines, such as interleukin (IL)-2, tumor necrosis factor (TNF)-α, and interferon (IFN)-γ; both those processes eventually impair the cytotoxicity of T cells to tumor cells [1,5,229,230]. Therefore, T cell exhaustion or T cell dysfunction can be a major barrier in the development of T cell-based or checkpoint therapy [76,234]. T cells that infiltrate glioblastomas tend to promote T cell exhaustion, as measured by the expression of immune checkpoints and decreased effector function, compared with T cells isolated from the peripheral blood of patients with glioblastoma, and that exhaustion increases adaptive immune resistance [136,234]. Tumor infiltrating lymphocytes (TILs) isolated from glioblastoma patients with a high percentage of exhausted T cells did not respond to anti-PD-1 inhibitors ex vivo [154].
NK cells are effector cells of the innate immune system. They recognize tumor cells by detecting the presence of receptor ligands that are upregulated in tumor cells, and then they target those cells without using the MHC; they also bind to tumor-specific antibodies secreted by B cells, which bind to antigens on the surfaces of tumor cells [183], and kill tumor cells directly. NK cells without tumor specificity could be efficacious for immunologically treating glioblastomas, which have high antigenic heterogeneity and a low mutational burden. NK cells can also be reproduced ex vivo. Restoring the exhausted T cell response and the limited cytotoxicity of endogenously activated T cells induced by vaccines can be achieved by using adoptively transferred T or NK cells, including tumor-specific antigen loading on ex vivo expanded T cells and genetically engineering T or NK cells to express chimeric antigen receptors (CARs). Moreover, modern advanced gene-editing technologies enable these effector cells to overcome immune escape mechanisms of tumor cells and relieve the immunosuppressive nature of glioblastoma microenvironment, which not only improve their antitumor immunity but also make them close to ideal off-the-shelf products.
Here, we provide an updated summary and discuss future directions for T and NK cell transfer therapies in glioblastoma.
ADOPTIVE T CELL THERAPY
Adoptive T cell therapy provides patients with a large number of immune effector cells that have been primed by a particular antigen and expanded ex vivo. The cells can be administered locally into the brain tumor site or systemically. A critical step in efficiently stimulating the adaptive immune response is the identification of appropriate target antigens. Identified TAAs of glioblastoma include IL-13 receptor alpha 2 (IL-13Rα2), human epidermal growth factor receptor 2 (HER2), erythropoietin-producing hepatocellular carcinoma A2 (EphA2), survivin, tyrosinase-related protein 2, Wilms’ tumor 1, glycoprotein 100 (gp100), SRY (sex-determining region Y)-box (SOX) 2, SOX11, melanoma-associated antigen 1 (MAGE-A1), absent in melanoma 2, and cytomegalovirus (CMV) proteins. Epidermal growth factor type III variant (EGFRvIII), isocitrate dehydrogenase 1 R132H, and H3.3 K27M can be classified as tumor-specific antigens (TSAs), and they are frequently shared among specific patient subgroups [54,146]. Targeting TAAs can cause life-threatening events by means of on-target off-tumor effects and T cell responses against normal cells [155]. On the other hand, the remarkable heterogeneity of antigen expression by glioblastomas [251] can be a strong barrier to adoptive T cell therapy that targets only one TSA. Moreover, the antitumor immune responses induced by peptide vaccines or DC vaccines that target multiple antigens or tumor lysates might be too diluted to control tumors.
The recruitment of T cells is a key process in immune response. Peripherally infused T cells can enter the central nervous system (CNS) in patients with glioblastoma, but only at the later stages of tumor growth when the blood-brain-barrier (BBB) has been destroyed [185]. The question of where to administer is another important issue in the application of adoptive T cell therapy for brain tumors. Intracranial delivery of T cells into brain tumors has shown encouraging results in terms of safety and therapeutic efficacy, compared with systemic exposure [208]. The advantages of intracranial delivery are overcoming the BBB, increasing tumor infiltration, decreasing the number of T cells required, and minimizing systemic toxicity.
Lymphokine-activated killer (LAK) cells
Adoptive T cell therapy has developed from LAK cell therapy, which transfers a mixture of IL-2-activated T cells and NK cells obtained by culturing patients’ peripheral blood mononuclear cells (PBMCs) in the presence of IL-2 to patients with malignant glioma [67]. The main cytotoxic property of LAK cells is mediated by CD3-CD56+ NK cells rather than CD3+CD56- T cells [159]. However, their limited cytolytic activity due to a lack of tumor specificity and their IL-2-related toxicity, such as brain edema and aseptic meningitis, have prevented widespread use of this strategy [9,70].
NK cells, CD3-CD56+ lymphocytes, play a pivotal role in the innate immune response. They contribute to the antitumor immune response by modulating T cell activation through the regulation of DC maturation, as well as by directly eliminating tumor cells [215]. They frequently infiltrate glioblastomas, but their lytic activity is decreased by the abundant immunosuppressive mechanisms of tumor cells and their microenvironment. Tumor recognition and the elimination of tumor cells by NK cells can be markedly enhanced through the expression of genetically engineered CARs. NK cell therapy will be discussed later.
TILs
TILs are effector T cells that are thought to have tumor specificity because they are already present in the tumor. Ex vivo expanded TILs are apt to proliferate in vivo and show functional activity and trafficking to the tumor [186]. It has been very difficult, however, to expand TILs from tumor tissues in most cancers, including glioblastomas, except melanomas [8]. Although some clinical studies in patients with recurrent [162] or newly diagnosed malignant glioma [163] used autologous TILs expanded ex vivo from cells in the draining inguinal lymph nodes after inoculation with irradiated autologous tumor cells have demonstrated a partial radiographic response, no survival benefit for the patients has been found. These results indicate that gliomas undoubtedly present immunosuppressive obstacles to TIL therapy, and a drastic improvement is needed in TIL expansion and the maintenance of TIL function in the immunosuppressive microenvironment of gliomas.
Adoptive CD8+ T cells
Selecting the target antigens is an important step in the efficient induction of antitumor immunity in effector T cells. Human CMV has been verified as a contributing factor of glioma progression [32] and suggested as a therapeutic target [81]. CMV pp65 antigens are recognized by a high fraction of T cells [127]. Because they are expressed in most glioblastomas (>90%) but not in normal brain tissue [33], they have been in the spotlight as target antigens for glioblastoma immunotherapy. In the first clinical study of adoptive immunotherapy using CMV-specific T cells in patients with recurrent glioblastoma, the treatment was shown to be safe with minimal toxicity to patients. The median overall survival (OS) of 19 patients was >57 weeks, with a median progression free survival (PFS) of >35 weeks [187]. Although CMV-specific immunotherapy showed disease stabilization and prolonged PFS in some patients, no correlation between antigen-specific T cell frequency and clinical outcomes was detected in that study. The tumor infiltrating CMV-specific T cells of some patients who showed tumor progression after T cell infusion displayed poor cytotoxic capacity and increased expression of inhibitory receptors such as PD-1, CTLA-4 and TIM-3, compared with T cells from peripheral blood. Moreover, regulatory T cells (Tregs) were detected at levels almost 5-fold higher in glioblastomas than in peripheral blood. Those disappointing results were ascribed to the infusion of CMV-specific CD8+ T cells without CD4+ helper T cells, the possible expansion of both Tregs and T cells in the presence of IL-2 loading, and including patients who were treated with T cells without prior lymphodepleting chemotherapy to make room for the infused T cells [223]. A phase I/II clinical trial using CMV pp65-specific T cells to remedy those limitations in patients with recurrent and newly diagnosed glioblastoma after lymphodepleting dose-dense temozolomide (TMZ) treatment, however, also found that the infused cells had insufficient cytotoxic function to control glioblastoma [223]. CMV-specific T cells might kill autologous tumor cells effectively in only a subset of CMV seropositive patients, which suggests that heterogeneity in CMV antigen expression could attenuate the effector function of T cells. Furthermore, the finding that CMV-specific T cells within the glioblastoma microenvironment were immunologically dysfunctional indicates that the tumor microenvironment (TME) of glioblastomas might be exceptionally immunosuppressive. Therefore, additional modulation will be needed to obtain effective antitumor immune responses to adoptive T cell therapy that targets CMV pp65.
In another clinical trial (NCT00693095) that tested CMV-targeting T cells with TMZ in 17 patients with newly diagnosed glioblastoma, an additional vaccination with CMV pp65 RNA-loaded DCs enhanced the frequency of polyfunctional CMV pp65-specific T cells after adoptive T cell therapy, correlating with prolonged OS [172]. Isolating CMV-specific T cells from glioblastoma patients with deficient polyfunctionality and then stimulating them with antigenic peptides in the presence of the γC cytokine ex vivo might reverse their inability to generate multiple cytokines and improve their ability to mount an effective antitumor response in vivo [35]. Repeated endogenous antigenic stimulation to adoptively transferred CMV pp65-specific T cells via DC vaccination seems to restore T cell polyfunctionality. The adoptive T cell therapy + DC vaccine platform can target tumor ribonucleic acid (RNA) instead of CMV pp65. Clinical trials to evaluate the safety and antitumor immune response of a tumor RNA-loaded DC vaccine and subsequent tumor RNA-specific T cell therapy in pediatric patients with newly diagnosed high-grade gliomas (NCT03334305) and patients with diffuse intrinsic pontine glioma (DIPG) (NCT03396575) are underway.
Genetically modified T cells
Genetic modification of T cells has been developed to enhance the antitumor efficacy of adoptive T cell therapy. Two approaches have commonly been used for this strategy : (a) transfer of complementary deoxyribonucleic acids in the α and β chains of the TCR cloned from high affinity TAA-specific T cells, and (b) insertion of CARs that recognize tumor cells through a single-chain variable fragment (scFv) isolated from TAA-specific antibodies [31,115].
TCR-transduced T cells
Genes encoding the TCRs of T cells isolated from patients can be transferred to the T cells of other patients with matching human leukocyte antigen (HLA) restrictions by cloning them into viral vectors [82]. In clinical studies of patients with metastatic melanoma who were treated with TCR-transduced T cells targeting melanoma-associated antigen recognized by T cells-1 (MART-1) or gp100 after lymphodepletion, objective cancer regression was seen in 30% (MART-1 targeting TCR) and 19% (gp100 targeting TCR) of patients who received human or mouse TCRs, respectively [82,138]. However, severe on-target off-tumor toxicity, including the destruction of normal melanocytes throughout the body (skin, eye, and ear) caused by cytotoxic T lymphocyte responses to cognate antigen-containing cells was observed. Moreover, two of nine patients with metastatic cancers that express the MAGE-A3 antigen, such as melanoma, synovial sarcoma, and esophageal cancer, who received TCR-transduced T cell therapy targeting MAGE-A3 died with severe brain damage from necrotizing leukoencephalopathy. MAGE-A3, which was not before known to exist in the brain, was later found there [137]. Another study to test the antitumor efficacy of autologous T lymphocytes genetically engineered to express a murine TCR against a human carcinoembryonic antigen in patients with metastatic colorectal cancer refractory to standard treatments was stopped when all three patients experienced life-threatening inflammatory colitis and colonic hemorrhage [155]. Those results indicate that genetically engineered TCR-transduced T cell therapy can induce the powerful destruction of normal cells that express the same antigens as the tumor cells. Perhaps because of those results, no clinical study of TCR-transduced T cell therapy has been performed in patients with glioblastomas, which has a paucity of TSAs. This approach has the further limitation that T cells engineered by this procedure generally recognize only antigens that have been processed and presented in an MHC-restricted pattern.
CAR T cells
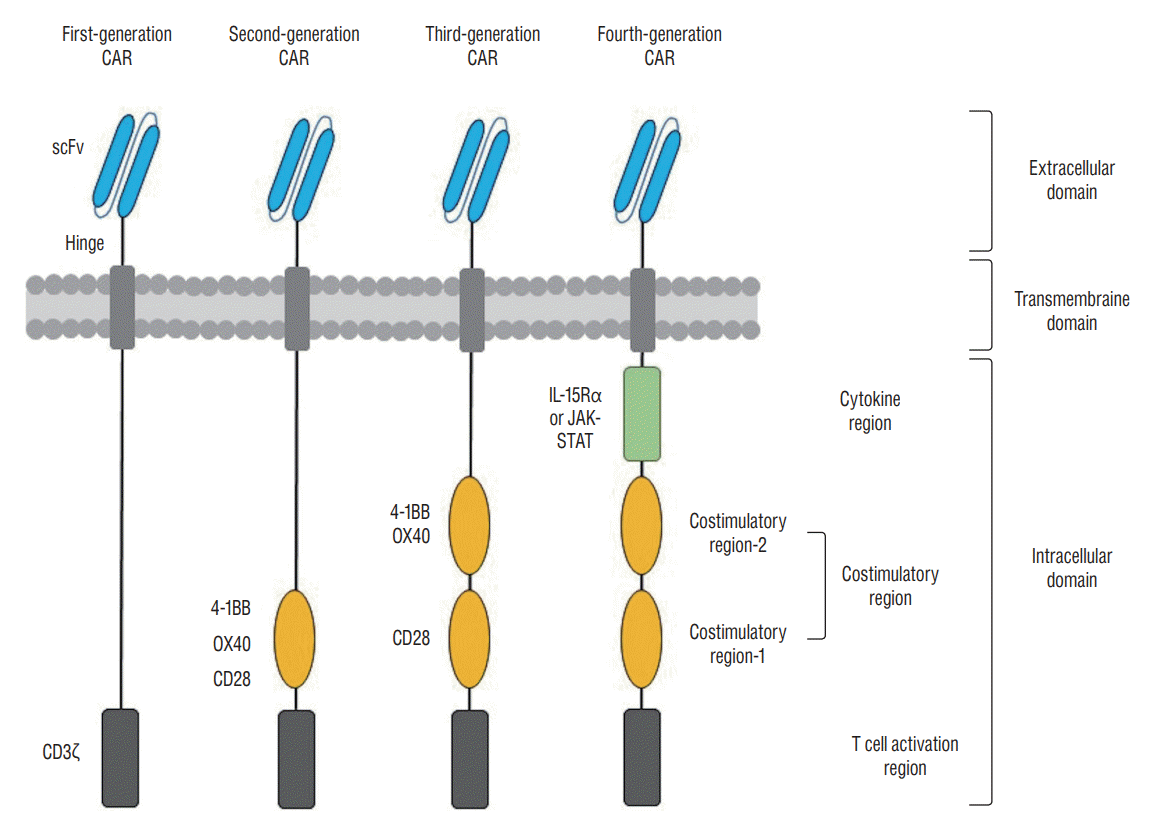
CAR T cell therapy refers to the adoptive transfer of T cells genetically modified to express CARs, recombinant molecules typically composed of an extracellular domain of a tumor antigen recognition molecule that contains the scFv of a monoclonal antibody, intracellular domains with a TCR signaling domain and an additional costimulatory domain that lead to T cell activation, and a transmembrane domain as a spacer [51,98]. The intracellular domain has been optimized in successive generations of CAR T cells to enhance its signaling capacity (Fig. 1). First-generation CARs that used only the CD3ζ chain as the intracellular activation domain demonstrated limited persistence and efficacy because they lacked costimulatory signals [16]. Second- and third-generation CARs were developed by combining the CD3ζ of the first-generation CARs with one (second-generation) or more (third-generation) costimulatory domains, such as CD28 and OX40 or 4-1BB [73,88]. The addition of cytokine signaling domains such as IL-15Rα or Janus kinase-signal transducers and activators of transcription into the intracellular domain of third-generation CARs produced fourth-generation CARs and enhanced the antitumor activity of third-generation CAR T cells [84,144]. Modified CAR T cells can recognize and kill target cells that express TSAs without the need for MHC presentation and costimulatory signals. In spite of the successful engineering of more potent and immunogenic CAR T cells, on-target off-tumor effects, poor tumor infiltration, and a highly immunosuppressive TME remain serious barriers to the clinical efficacy of CAR T cells for solid tumors, including glioblastomas [169].
It is also important to select appropriate antigens to target in CAR T cell therapy. An ideal target antigen is homogenously expressed on all tumor cells and completely absent on normal cells. To prevent normal cell attack by CAR T cells, which have greater cytotoxicity than non-engineered T cells, tumor antigens should be undetectable or have minimal expression on normal tissues not enough to mediate the elimination of normal cells. It is very important, therefore, for phase I clinical trials in patients with glioblastomas that mainly express TAAs to confirm the safety and feasibility of this treatment. Antigens selected for CAR T cells in glioblastomas to date include IL-13Rα2, EGFRvIII, HER2, and EphA2. To overcome the antigenic variability of glioblastomas and increase the efficacy of CAR T cells, bi- and tri-specific CAR T cells that target multiple TAAs have been developed and shown increased antitumor efficacy [12,71]. Ongoing clinical trials of CAR T cell therapy for glioblastoma are summarized in Table 1.
EGFRvIII
EGFRvIII, a mutated form of EGFR, is expressed in about 30% of glioblastomas [231]. EGFRvIII might be a potentially ideal target for CAR T cell therapy because its extracellular epitope can easily be recognized by monoclonal antibodies, and this mutation is absent in normal tissues. EGFRvIII-specific CAR T cell therapy has shown effective tumor control in preclinical studies [182], but it has had limited success in clinical trials [147]. In a phase I clinical trial that used a single infusion of autologous second-generation CAR T cells (CD3ζ + CD28) targeting EGFRvIII in ten recurrent glioblastoma patients without prior lymphodepletion, all patients had detectable engraftment of EGFRvIII CAR T cells in their peripheral blood [147]. The EGFRvIII CAR T cells successfully trafficked into the tumors in the brain, with antigen decrease, indicative of antigen-specific tumor cell lysis, in five of seven patients who received surgery early after the infusion. Some of the tumor specimens, however, had infiltration of immunosuppressive Tregs and increased expression of inhibitory molecules such as indoleamine 2,3-dioxygenase 1, programmed deathligand 1 (PD-L1), and IL-10. Those results suggest that EGFRvIII CAR T cell therapy might not only induce antigen-specific antitumor cytotoxicity, but also stimulate a compensatory immunosuppressive response. In another phase I clinical trial, third-generation EGFRvIII CAR T cells (CD3ζ + CD28 and 4-1BB costimulatory domains) were administered after lymphodepleting chemotherapy and supported post-transfer with intravenous IL-2 in patients with recurrent glioblastoma [64]. Eighteen patients were treated intravenously with a dose escalation of EGFRvIII CAR T cells. The persistence of CAR T cells correlated with the cell dose, but no objective antitumor responses were observed.
HER2
HER2 is another member of the epidermal growth factor receptor family that plays an essential role in cell proliferation, differentiation, motility, and adhesion, so its overexpression in cancer is usually associated with a poor prognosis [97]. HER2 is expressed at high levels (about 80%) in glioblastomas [134], but it is also present in normal cells; therefore, HER2 CAR T cells could pose the risk of on-target off-tumor toxicity. Such autoimmune toxicity was manifest in a patient with metastatic colon cancer treated with third-generation (CD3ζ + CD28 and 4-1BB) ErbB2 (HER2/neu) CAR T cells and IL-2 after lymphodepleting chemotherapy that died of respiratory distress due to a cytokine storm triggered when the ErbB2 CAR T cells recognized low levels of ErbB2 on lung epithelial cells [139]. That case emphasizes the importance of selecting TAAs with limited expression on normal cells because CAR T cell therapy has potent cytotoxicity. Nonetheless, in a subsequent phase I study, autologous HER2 CAR T cells (CD3ζ + CD28) were successfully demonstrated to be safe without dose-limiting toxicity in 17 patients with progressive glioblastoma [2]. Major differences between the first troubling case and the latter study were the use of a second-generation CAR (CD3ζ + CD28) with a different scFv (FRP5-based exodomain), the absence of a concomitant IL-2 infusion, and no use of prior lymphodepleting chemotherapy. The HER2 CAR T cells persisted in the peripheral blood for up to 12 months. Eight patients had clinical benefits, in the form of a partial response (n=1) or stable disease (n=7). The median OS was 11.1 months after T cell administration and 24.5 months after diagnosis.
IL-13Rα2
IL-13Rα2, a cancer-germline antigen found in both glioma cells and the testes, is not expressed at significant levels in normal brain tissue [40,87]. The activation of IL-13Rα2 is associated with increased invasiveness of glioblastoma, so IL-13Rα2 overexpression is related to poor prognosis [20]. Local administration of first-generation IL-13Rα2 CAR T (CD3ζ) cells into the resection cavity of three patients with recurrent glioblastoma was found to be feasible and safe, with encouraging clinical responses, in a first-in-human pilot study [18]. A subsequent study of serial intracranial and intraventricular infusions of second-generation CAR T cells targeting IL-13Rα2 (CD3ζ + 4-1BB) in one patient with multifocal glioblastoma also showed the treatment to be safe and feasible. All intracranial and metastatic tumors in the spine were completely eliminated during the treatment, and that dramatic clinical response was sustained for 7.5 months after the initiation of therapy (NCT02208362) [17]. A phase I clinical trial (NCT05540873) to assess the safety and tolerability of intravenously administered IL-13Rα2 CAR T cells in patients with malignant glioma has started in Korea in September 2022.
B7 homolog 3 protein (B7-H3)
B7-H3, also known as CD276, is a member of the B7 superfamily of immune checkpoint molecules that regulate T cells [34]. It is overexpressed in the cells of some hematological and most solid tumors [135], and it shows limited expression in normal tissues [212]. Therefore, B7-H3 could be an attractive target for antibody-based cancer immunotherapy. It is expressed, interestingly, in tumor-associated vessels and fibroblasts as well. Thus B7-H3 CAR T cells could eliminate tumor cells not only through direct targeting, but also through stroma disruption and neo-angiogenesis inhibition [160,188] without causing major on-target off-tumor toxicity. Second-generation (CD3ζ + 4-1BB) B7-H3 CAR T cells effectively controlled solid tumor cells [47] and induced the regression of established solid tumors in xenograft models of osteosarcoma, medulloblastoma, and Ewing’s sarcoma without causing obvious toxicity [125]. A randomized, parallel-arm, phase I/II study (NCT04077866) of an intratumoral/intraventricular injection of B7-H3 CAR T cells between TMZ cycles to evaluate their safety and efficacy in patients with refractory or recurrent glioblastoma is in progress.
CD147
CD147, an extracellular matrix (ECM) metalloproteinase inducer, is one of the immunoglobulin superfamily of adhesion molecules that stimulate collagenase secretion, such as metalloproteinase-1, -2, -3, -9, -14, and -15 in fibroblasts, that leads to the degradation of the ECM [237]. It is overexpressed in tumor cells, including glioblastoma cells, and is believed to increase their malignant properties, such as proliferation, invasion, metastasis, and the inhibition of tumor cell apoptosis [66,102]. In addition, it might also be involved in angiogenesis via the regulation of vascular endothelial growth factor production in tumor and stromal cells [14]. Therefore, it is a promising biomarker for predicting prognosis in many cancers, including glioblastomas [53,241]. A single-center, single-arm, open label, dose-escalation clinical study (NCT04045847) to assess the safety, tolerance, and efficacy of CD147 CAR T cells is underway in patients with recurrent glioblastoma.
Disialoganglioside (GD2)
GD2 is highly expressed on several types of tumor, including melanoma, retinoblastoma, and neuroblastoma [120]. It is also highly expressed in glioblastoma cells and expressed at a very low level in normal CNS cells [46]. The antitumor effects and safety of GD2 CAR T cells have been explored in various preclinical models, including glioblastoma [58,167] and diffuse pontine glioma [140]. A clinical trial (NCT04196413) to evaluate the antitumor efficacy of GD2 CAR T cells after lymphodepleting chemotherapy in patients with H3-K27M mutated diffuse pontine glioma is ongoing. In a preliminary report, three of the first four patients exhibited clinical and radiographic improvement without on-target off-tumor toxicity [124]. Another phase I clinical study (NCT04099797) of GD2 CAR T cell therapy in patients with GD2-expressing brain tumors, such as glioblastoma, DIPG, medulloblastoma, and other rare brain tumors, is also underway. The insertion of GD2-targeting CARs into mesenchymal stem cells (MSCs) that deliver TNF-related apoptosis-inducing ligand (TRAIL) to create CARs with bi-functional MSCs that express high levels of both TRAIL and GD2 could reinforce the antitumor activity of this treatment against GD2-positive glioblastoma cells [65].
Matrix metalloproteinase 2 (MMP-2)
Another way to expand the repertoire of target antigens used in CAR T cell therapy involves naturally derived products with exclusive tumor-binding potential [170]. For example, chlorotoxin (CLTX), a peptide toxin isolated from the venom of the death stalker scorpion (Leiurus quinquestriatus) [39], can selectively bind to glioblastoma and other neuroectodermal tumors, while showing minimal reactivity with normal cells in the brain and other tissues, including skin, kidney, and lung tissues [121,195]. CLTX itself is non-cytotoxic to tumor and normal tissues, so it has been used for the tumor-specific delivery of cytotoxic agents or radioisotope I [131 38,126]. Although the precise cell surface receptor on glioblastoma cells that is responsive to CLTX remains unclear, the expression of MMP-2, chloride channel CLCN3, and phospholipid protein annexin A2 seems to be involved [43,129,201]. Specific binding of CLTX to cancer cells is facilitated by MMP-2 [210], and MMP-2 knockdown in glioblastoma cells reduced CLTX CAR T cell activation and cytotoxicity [218]. Thus, MMP-2 expression is required for effective CLTX CAR T cell activation. CLTX CAR T cells also efficiently eradicated tumors in glioblastoma-bearing mice with no observed toxicity [218]. A subsequent phase I study (NCT04214392) of T cells expressing CLTX CARs for the treatment of MMP-2-positive recurrent or progressive glioblastoma is in progress.
EphA2
EphA2, a member of the Eph family of receptor tyrosine kinases, is overexpressed in glioblastomas [236] and associated with poor outcomes [221] through its capacity to enhance tumorigenesis [248], tumor invasion [132], angiogenesis [45,149], and metastasis15). EphA2 is not expressed in most normal tissues, including the brain, but it is present in pulmonary epithelial cells [236]. Second-generation EphA2 CAR T cells (CD3ζ + CD28) could eliminate EphA2-positive glioblastoma cells and glioblastoma-initiating cells in vitro, and adoptive transfer of EphA2 CAR T cells in animal models showed the regression of gliomas and a significant survival benefit [30]. In a subsequent study, incorporating the 4-1BB signaling domain into CD3ζ + CD28 CARs did not improve CAR T cell function, so second-generation CAR T cells (CD3ζ + CD28) might be a safer choice for clinical trials than third-generation CAR T cells [244]. A pilot study (NCT03423992) is in progress to determine the safety and efficacy of personalized CAR T cell immunotherapy based on the expression of TSAs/TAAs (EGFRvIII, IL13Rα2, HER2, EphA2, CD133, GD2) in patients with recurrent malignant gliomas. In the preliminary report, two of three patients with EphA2-positive recurrent glioblastoma enrolled as the first cohort to receive a single intravenous infusion of EphA2 CAR T cells at a starting dose level of 1×106 cells/kg after lymphodepleting chemotherapy showed grade 2 cytokine release syndrome (CRS) accompanied by pulmonary edema, which resolved with dexamethasone treatment. Among those three patients, one achieved stable disease, and the other two patients showed progressive disease, with OS ranging from 86 to 181 days [114].
NK group 2 member D (NKG2D) ligands
The human NKG2D is an activating receptor naturally expressed on most NK cells, CD8+ T cells, a subset of CD4+ T cells, NK T cells, and γδ T cells. Cells undergoing stress such as DNA damage, hypoxia, or viral infection can express NKG2D ligands [11]. The interaction between NKG2D and NKG2D ligands causes immune cell activation that results in the cytolysis of NKG2D ligand-expressing cells [13]. NKG2D ligands are rarely expressed by normal tissues, but they are frequently overexpressed on solid tumors [145]. NKG2D ligands are also frequently expressed on glioblastoma stem-like cells and glioblastoma cells, and they can activate NKG2D-expressing killer cells [55,239]. Chemotherapy or radiotherapy can upregulate NKG2D ligand expression on glioblastoma cells, so combining conventional therapy with NKG2D-targeting immunotherapy might have synergistic antitumor effects [225]. Such an effect, including significantly prolonged OS, was actually demonstrated in an animal study of glioblastoma combining murine NKG2D CAR T cell therapy with radiotherapy [226]. A phase I study (NCT04270461) to evaluate the safety and clinical activity of NKG2D-based CAR T cells in the treatment of relapsed and refractory NKG2DL-positive solid tumors, including glioblastoma and medulloblastoma, was withdrawn for administrative reasons. Recently, increased antitumor activity of human mRNA-based multifunctional NKG2D CAR T cells coexpressing IL-12 and IFN-α2 was reported in vitro and in vivo in mouse glioma models without signs of toxicity [131].
In addition to those targets of CAR T cell therapy in ongoing clinical studies, targets in preclinical studies are carbonic anhydrase IX (CAIX), CD70, chondroitin sulfate proteoglycan 4, fibroblast growth factor-inducible 14 (Fn14), and trophoblast cell surface antigen 2.
ENHANCEMENT OF CAR T CELL FUNCTION
Targeting multiple antigens
The antigenic and molecular profiles of glioblastoma are strikingly heterogeneous in terms of pathology and genetic changes, even within a single tumor [156]. So, glioblastoma cells without the targeted antigen can escape CAR T cell recognition and elimination. In addition, preclinical and clinical studies have shown that targeting a single antigen can result in antigen loss variants during subsequent tumor recurrence [99,150]. Strategies to prevent such escape include efforts to expand the list of available TSAs, such as mutation-derived neoantigens, and to engineer CAR T cells to achieve multispecificity. To date, two preclinical studies from the same research group have used CAR T cells to target multiple antigens in glioblastomas. One study used bispecific CARs composed of signaling domains (CD28 + CD3ζ) and a tandem CAR (TanCAR) exodomain that fused a HER2-binding scFv to an IL-13Rα-binding IL-13 mutein. The TanCAR T cells displayed enhanced antitumor efficacy and improved animal survival compared with the effects of single CAR T cells encountering HER2 or IL-13Rα2 [71]. The other study designed trivalent CAR T cells that targeted HER2, IL-13Rα2, and EphA2. The trivalent CAR T cells exhibited improved cytotoxicity and cytokine release compared with monospecific and bispecific CAR T cells in vitro. They were also able to control tumor growth at low T cell doses in autologous glioblastoma patient-derived xenografts [12]. Further clinical information is required to determine whether CAR T cells targeting multiple antigens can be efficient in the immunosuppressive TME of human glioblastomas.
Neoantigens
Neoantigens are real TSAs because they are a series of peptides present in tumor cells but not in normal cells. Therefore, neoantigens derived from tumor-specific mutations can generate potent immune responses with central tolerance and without toxicity to normal tissue [242]. The most common method for identifying personalized neoantigens compares DNA sequences in tumor tissues with those in normal tissue [108]. An efficient sequencing tool currently in wide use is whole exome sequencing technology [209].
Since the first personalized neoantigen-pulsed DC vaccine began to be tested in a phase I clinical trial for melanoma in 2015 [22], various clinical trials of neoantigen-loaded DC vaccines for solid tumors have been conducted [44,151,152,180,184] and shown therapeutic value against cancer. In a phase I/Ib study of personalized neoantigen DC vaccines for eight patients with newly diagnosed O6-methylguanine-DNA methyltransferase (MGMT)-unmethylated glioblastoma, neoantigen-specific T cells from the peripheral blood were shown to migrate into an intracranial tumor without dose-limiting toxicity. However, all patients showed tumor recurrence and ultimately died of progressive disease, with a median OS and PFS of 16.8 months and 7.6 months, respectively [89]. Thus, even the T cell response induced by a DC vaccine targeting neoantigens might be insufficient to produce clinically effective antitumor activity in the immunosuppressive TME of glioblastoma. Perhaps, reducing the number of steps between effector T cells and naïve T cells in the body would improve the antitumor activity of effector T cells. For example, in mouse tumor models, neoantigen-pulsed DC vaccines that used fewer steps to produce effector T cells endogenously were superior to neoantigen-adjuvant vaccines with one more step of antigen priming in both activating immune responses and inhibiting tumor growth [252]. Therefore, ex vivo expanded neoantigen-specific effector T cells that do not require an endogenous process to stimulate T cells might improve the antitumor immune response.
Cytokine overexpression
Incorporating a signal of immune stimulatory cytokines into third-generation CAR T cells produces fourth-generation CAR T cells, which have improved cell expansion and persistence [25]. Transgenic cytokine expression could also stimulate CAR T cells to lyse antigen-negative cancer cells not otherwise recognized by CAR T cells in the tumor. The cytokine tested in a glioblastoma animal model was IL-15, which plays an important role in T cell expansion and survival, especially in absence of the antigen [92,99]. IL-13Rα2 CAR T cells modified to express transgenic IL-15 (IL-13Rα2 CAR T/IL-15) showed greater proliferation and longer persistence, and produced more cytokines than IL-13Rα2 CAR T cells in vitro. Those results also produced a survival benefit in vivo, but late recurrence of tumors with downregulated IL-13Rα2 expression and antigen loss was observed [99]. Although genetically modifying CAR T cells to express transgenic cytokines might be a powerful method to improve their antitumor activity, multiple antigen targeting might also be required to prevent the occurrence of antigen loss variants during tumor recurrence.
Bispecific T cell engagers (BiTEs)
BiTEs are a subclass of bispecific antibody composed of two different antibody fragments, one of which is specific for CD3ζ on T cells and the other of which recognizes a tumor antigen [233]. Therefore, BiTEs can act as an immunologic synapse to facilitate an optimal interaction between cytotoxic T cells and tumor cells without the need for co-stimulation or MHC recognition [48]. Their antitumor effects and safety have been shown in clinical trials for various hematologic malignancies [10,143] and solid tumors [94,200]. In glioblastoma mouse models, bispecific T cells expressing both EGFRvIII CAR and a BiTE against EGFR eliminated heterogeneous EGFRvIII-expressing tumors that monospecific EGFRvIII CAR T cells were unable to treat completely [28]. Fn14, a cell surface receptor of the TNF-related weak inducer of apoptosis, is upregulated in gliomas, and its overexpression can stimulate the migration and invasion of glioma cells, so it is associated with poor prognosis [206]. In a preclinical study testing the antitumor activity of three therapeutic approaches, an Fn14×CD3ζ BiTE antibody, Fn14-specific CAR T (Fn14 CAR T) cells, and Fn14 CAR T cells engineered to secrete IL-15 (Fn14 CAR T/IL-15) against glioblastomas, both the Fn14xCD3ζ BiTE antibody and Fn14 CAR T cells showed cytotoxic effects in vitro and in vivo, and IL-15 production augmented the antitumor effects of CAR T cells, resulting in longer remission and survival [106]. Although BiTEs or BiTE-secreting CAR T cells have shown promising antitumor activity in preclinical studies, it is necessary to further verify whether they will work well in the immunosuppressive TME of human glioblastomas. A phase I clinical study (NCT04903795) to evaluate the safety of EGFRvIII×CD3ζ BiTEs alone and in combination with a peripheral autologous T cell infusion in patients with EGFRvIII-mutated grade IV malignant glioma is ongoing.
Disrupting immunosuppressed molecules
The widespread adoption of gene-editing technologies has enabled the random insertion or deletion of specific transgenes to or from CAR T cells. Incapacitating immune inhibitory molecules such as PD-1 and transforming growth factor-β (TGF-β) on T cells could be one strategy for overcoming the immunosuppressive TME of glioblastomas.
Methods for blocking or reversing immune inhibitory molecules include combination therapy of CAR T cells and PD-1 blocking antibodies [190,194], CAR T cells that secrete PD-1 blocking antibodies [198], CAR T cells with PD-1 gene-knockout [29], and CAR T cells with a PD-1 chimeric switch receptor that reverses the inhibitory signal of PD-1 activation into a stimulatory signal [116]. Because Treg cells also express PD-1, systemic treatment with PD-1/PD-L1 blocking agents could enhance Treg cell function, leading to significant suppression of antitumor immune responses and subsequent hyper-progression of cancers [23,90]. The advantages of CAR T cells with an intrinsic PD-1 blockade produced by genetic engineering are that they provide more sustainable activity and more tumor-limiting PD-1 inhibition than CAR T cells combined with antibody treatment. Targeted disruption of PD-1 using the clustered regularly interspaced short palindromic repeats-CRISPR associated protein 9 (CRISPR-Cas9) system enhanced the antitumor activity of EGFRvIII CAR T cells in vitro and significantly prolonged the survival of mice bearing glioblastomas [29]. However, a sustained PD-1 blockade by genetic deletion can promote the accumulation of terminally differentiated, exhausted CD8+ T cells [148,224] and raise safety concerns about the occurrence of CRS through the supraphysiological activation of CAR T cells that would result from continuously blocking the physiologic function of PD-1, which is to inhibit excessive T cell activation. Clinical trials, therefore, should only be conducted after sufficient consideration of the problems that could arise from a persistent PD-1 blockade.
TGF-β is a powerful immunosuppressive factor that has been shown to promote T cell exclusion and dysfunction in most solid tumors, including glioblastomas [128,161]. TGF-β blockade can facilitate the efficacy of adoptive T cell therapy for glioblastomas. Recent advanced CAR engineering can convert immunosuppressive molecules into T cell stimulants for CAR T cell therapy. Actually, CARs responsive to a variety of soluble ligands, including TGF-β, can be constructed to effectively convert TGF-β from a potent immunosuppressive cytokine into a strong stimulant for T cells [24]. Such CAR T cells could inhibit endogenous TGF-β signaling in T cells. Because that approach is not directly involved in tumor cell lysis, it might require a combination of receptors to recognize surface antigens for direct tumor cell killing.
Enhancing T cell trafficking
Making CAR T cells accumulate in the TME for a long time is another method for increasing antitumor efficacy. CAR T cells can be engineered to express chemokine receptors, such as C-X-C motif chemokine receptor 1 (CXCR1) and CXCR2, and thereby enhance intratumoral T cell trafficking. CD70, a member of the TNF family, is a novel immunosuppressive ligand and glioma target [79]. CD70 CAR T cells modified to express IL-8 receptors, CXCR1, and CXCR2 had greater trafficking to the tumors via radiotherapy-induced IL-8 upregulation, which resulted in complete tumor regression and a long-lasting memory T cell response in preclinical models of malignant tumors, including glioblastoma [80]. A subsequent phase I study (NCT05353530) of IL-8 receptor-modified CD70 CAR T cell therapy in CD70 positive and MGMT-unmethylated adult glioblastoma is underway.
Allogeneic CAR T cells
CAR T cell therapy based on autologous T cells has some limitations, including treatment delay due to production time, high cost, and the risk of manufacturing failure, and the functional availability of T cells is often reduced by the disease itself or previous therapies [254]. Allogeneic or universal “off-the-shelf” CAR T cell therapy using T cells obtained from healthy donors could be an alternative. Advances in gene-editing technologies, including zinc finger nuclease (ZFN) [168], transcription activator-like effector nuclease [164], and CRISPR/Cas9 [174], might be used to remove the two main barriers to allogeneic CAR T cells : graft-versus-host disease (GVHD) and allorejection. These exquisite gene-editing tools can generate TCR-deficient T cells to prevent GVHD [164,174,204] or T cells that eliminate MHC class I molecules by disrupting the β2-microglobulin locus to reduce allorejection [217]. In a recent phase I clinical trial, the safety and feasibility of allogeneic CAR T products were evaluated in patients with recurrent glioblastomas. Healthy donor-derived CAR T cells targeting IL-13Rα2 were generated and engineered using ZFNs to permanently disrupt the glucocorticoid receptor GRm13Z40-2, making them resistant to glucocorticoid treatment. Allogeneic Grm13Z40-2 T cells combined with an intracranial infusion of IL-2 and systemic dexamethasone maintained their effector function in the presence of dexamethasone, which was used to reduce the tumor-related brain edema as well as the rejection of therapeutic allogeneic T cells, and induced transient tumor reduction and/or tumor necrosis at the T cell infusion site in four of six treated patients without evidence of GVHD [19]. This first-in-human experience demonstrated the feasibility of using allogeneic CAR T products to treat glioblastomas.
ADOPTIVE NK CELL THERAPY
NK cells are effector cells of the innate immune system that can kill tumor cells directly in an MHC-independent fashion by releasing lytic granules that contain perforin and granzymes or by inducing death receptor-mediated apoptosis through the expression of the Fas ligand or TRAIL [166]. The mechanism of NK cell activation is subject to the “missing-self hypothesis”. NK cells do not attack healthy cells when their inhibitory receptors, including NKG2A and killer immunoglobulin-like receptors (KIR), recognize the cognate MHC class I molecules of healthy cells, which protects self-cells from innate immunity; however, the downregulation of MHC class I occurs frequently in tumor cells, which are thus subject to NK cell activation [100,118]. NK cells recognize tumor cells through cell surface receptors such as NKG2D, CD16, and the natural cytotoxicity receptors NKp44, NKp46, and NKp30, which bind directly to ligands of the tumor cells [119]. Those receptors activate signaling proteins such as DAP10, DAP12, and CD3ζ that initiate the release of perforin and granzymes, and mediate the release of cytokines such as IFN-γ and TNF-α, resulting in the lysis of tumor cells [103]. NK cells can also regulate DC maturation through crosstalk with DCs that determines the efficacy of the DC-mediated adaptive immune response [50,216]. Furthermore, NK cells can eliminate tumor cells via antibody-dependent cellular cytotoxicity (ADCC) mediated by CD16 [105]. They express Fcγ receptors that bind to tumor cell surface-coating tumor-specific antibodies secreted by B cells and then lyse tumor cells by releasing perforin and granzymes [213]. In addition, they infiltrate glioblastomas more frequently than T cells [240]. In immunotherapy for glioblastomas, innate immune NK cells without tumor specificity can have distinct advantages over adaptive immune T cells because glioblastoma presents high antigenic heterogeneity and a low mutational burden. Therefore, NK cells could be potential candidates for adoptive immunotherapy for glioblastoma, if their potent cytotoxicity can be maintained in vivo despite the severely immunosuppressive TME and tumor cells.
Ongoing clinical trials of allogeneic and CAR NK cells are summarized in Table 2.
Allogeneic NK cells
Allogeneic HLA-mismatched NK cells have been in the spotlight as an alternative to autologous cells. Because allogeneic NK cells can bypass inhibitory signals and carry a low risk of GVHD [158,178], they are expected to have more potent antitumor efficacy than autologous cells. Moreover, glioblastoma stem-like cells seem to be highly susceptible to the cytotoxicity of allogeneic NK cells [6,69]. The sources of allogeneic cells include embryonic stem cells (ESCs), induced pluripotent stem cells (iPSCs) [49], umbilical cord blood [211], cell lines such as NK-92 [93], and the PBMCs of healthy donors [133]. Because of the low yield of NK cells available from allogeneic sources and the low transduction efficiency of CAR constructs, it could be reasonable to use NK cell lines. The NK-92 cell line is approved by the United States Food and Drug Administration (US FDA) for use in clinical trials [202]. NK-92 cells can expand without limit and are uniform, well-characterized, reproducible, and easily modifiable through genetic engineering [192,199]. They present impaired ADCC due to a lack of CD16, which can be re-expressed by CRISPR/Cas9 editing to increase antitumor cytotoxic activity [75]. Because the NK-92 cell line is derived from human NK cell lymphoma, it requires irradiation prior to infusion into patients due to safety concerns, such as chromosomal abnormalities and the risk of malignant transformation [175]. This radiation treatment does not affect the cytotoxicity of NK-92 cells, but it impairs their proliferation and reduces trafficking to the tumor, so it does limit their therapeutic efficacy [72]. Therefore, CAR NK-92 cells must be administered several times. Allogeneic NK cell transfer therapy has also been found to be safe. The antitumor efficacy of allogeneic NK cell therapy has been demonstrated in treating hematologic malignancies and to a lesser extent in studies on solid tumors [60]. The US FDA approved a clinical trial (NCT04489420) to evaluate the safety and feasibility of using human placental hematopoietic stem cell-derived NK cells (CYNK-001) in patients with recurrent glioblastoma in July 2020, but unfortunately that study was terminated in January 2022.
CAR NK cells
Because NK cells have potent antitumor cytotoxicity and a favorable safety profile, they are the most frequently explored candidate for generating CARs. NK cells do not produce IL-1 or IL-6, the main cytokines involved in CRS [192], and they display a low risk of GVHD [158,178]. If CARs are genetically engineered on NK cells, they will have greater antitumor activity than CAR T cells and minimal toxicity. The development of CAR NK cells for glioblastomas has followed a path similar to that of CAR T cells. CAR NK cells used CD3ζ as the first signal domain and then costimulatory domains such as CD28 and CD137 (4-1BB) were added. These conventional costimulatory domains, which are not found in NK cells [72], can be changed into NK-specific signaling domains such as NKG2D, CD244 (2B4), DAP10, or DAP12 to promote NK cell activation and cytotoxicity [62,130]. Compared with CAR T cells, CAR NK cells carry significantly fewer safety concerns, such as CRS and GVHD [158]. Moreover, CAR NK cells maintain their activating receptors, including NKp30, NKp44, NKG2D, and DNAM-1, which could reduce tumor recurrence caused by the loss of CAR targeting antigens [192]. The CAR targets of NK cells, including HER2, EGFR, EGFRvIII, and NKG2D, are very similar to those of T cells discussed above.
The potent anti-glioblastoma activity of CAR NK cells targeting EGFR [61], EGFRvIII [61,141,142], both EGFR and EGFRvIII [61,68], and ErbB2 (HER2) [4,249,250] has been shown in various preclinical studies. The route of delivery is also important in CAR NK cell therapy. Intravenously injected CAR NK-92 cells did not cross the BBB without ultrasound disruption in murine models, resulting in no therapeutic efficacy for intracranial tumors [4]. Even though the BBB environment in animals might be different from that in human glioblastoma patients whose BBB can be broken by the tumor, intratumoral delivery has been the preferred route of administration. In preclinical glioblastoma mouse models, repeated intratumoral injections of ErbB2 (HER2)-specific CAR NK-92 cells (NK-92/5.28.z cells) induced endogenous antitumor immunity and persistent protection against the tumor, with cures of initial syngeneic glioblastomas and the rejection of rechallenged tumor cells at distant sites [249]. A subsequent phase I clinical trial, CAR2BRAIN (NCT03383978), to investigate a clonal intracranial ErbB2-specific NK-92/5.28.z CAR NK product combined with intravenous ezabenlimab in patients with recurrent HER2-positive glioblastoma is ongoing.
Protection from an immunosuppressive microenvironment
NK cell therapy for glioblastoma can encounter several obstacles, including the inhibition of NK cell infiltration into tumor sites, downregulation of target ligands or maintenance of cognate MHC class I molecule expression on the tumor cells, and the release of inhibitory cytokines and secretory factors such as TGF-β in the TME [183]. TGF-β impairs NK cell cytotoxicity and proliferation by inhibiting IFN-γ [117], and it also inhibits activating receptors such as NKKG2D [104] and ADCC [207]. So blocking the TGF-β signaling pathway could be a strategy to increase NK cell function [238]. Genetically engineering cord blood-derived NK cells to express dominant negative TGF-β receptor II, a mutant receptor lacking the kinase domain of TGF-β, has shown enhanced antitumor activity in preclinical studies of glioblastoma [246] and medulloblastoma [165]. Another approach is administering TGF-β inhibitors with the NK cells. In a xenograft glioblastoma mouse model, treatment with allogeneic NK cells in combination with inhibitors of integrin or TGF-β signaling or treatment with allogeneic NK cells whose TGF-β receptor 2 gene was edited to abrogate glioma stem cell-induced NK cell dysfunction produced significant tumor control and prolonged survival of the animals [189]. Those findings suggest that the integrin and TGF-β axis could be a potential therapeutic target of NK cell therapy in glioblastomas, as well as an important NK cell immune escape mechanism. A phase I clinical trial (NCT04991870) evaluating the feasibility and toxicity of engineered allogeneic cord blood NK cells with TGF-βR2 and NR3C1 deletion in recurrent glioblastoma is in progress. Mothers against decapentaplegic homolog 3 (SMAD3) could be an another target for NK cell therapy. SMAD3 can induce TGF-β mediated NK cell suppression, so the suppression of SMAD3 could enhance NK cell activity [207]. Genetically engineered SMAD3-silenced NK-92 cells promoted IFN-γ production in NK-92 cells and inhibited tumor progression in xenograft mouse models of hepatoma and melanoma [222]. In addition, prostaglandin E2 (PG E2) secreted by cancer cells can promote cancer progression by inhibiting NK functions [153]. Blocking PG E2 has enhanced NK cell activity in preclinical models of metastatic breast cancer [123] and gastric cancer [110].
An approach targeting immune checkpoints can be applied to NK cells as well as T cells to improve their potential antitumor immunity. These immune checkpoints include NK cell-specific receptors such as KIR and NKG2A, and NK cell-expressed TIM-3, T cell immunoreceptor with immunoglobulin and immunoreceptor tyrosine-based inhibition motif domains, CD96, and LAG-3 [5,101]. Although the role of those checkpoints in regulating NK cell function remains unclear and the mechanisms involved in their enhancement of NK cell function are controversial [91], further studies evaluating the therapeutic benefits of targeting them will provide new treatment options and improve NK cell function, producing better clinical outcomes.
Off-the-shelf CAR NK cells
Off-the-shelf products in effector cell therapy are allogeneic immune cells that can be manufactured on a large scale and distributed to treat a broad range of cancer patients [193]. Although CAR NK-92 cells with potent antitumor activity do not require strict HLA matching, can expand easily, and do not present safety concerns such as GVHD and CRS, making them good candidates for off-the-shelf products, they require irradiation prior to infusion into patients, which suppresses their proliferation. Moreover, these cells do not express CD16, which mediates ADCC, so they might have decreased lytic activity. iPSCs might also be standardized as an off-the-shelf therapy. Human iPSCs can produce NK cells effectively [95], and they are easier to genetically modify than ESCs and hematopoietic stem cells [111]. Theoretically, any somatic cell can be reprogrammed into an iPSC, but in practice, easily accessible cells such as skin, urine, or blood are commonly used [85].
iPSC-derived NK cells expressing CARs that use NK-specific NKG2D instead of conventional CD28 as the costimulatory signal (NK-specific CAR iPSC NK cells) have demonstrated enhanced NK cell activation and longer survival than T cells or iPSC-NK cells expressing a conventional CAR in a mouse xenograft model of ovarian cancer [111]. NK-specific CAR iPSC NK cells could be an attractive option as a safe and renewable off-the-shelf CAR NK therapy, but a variety of clinical studies to evaluate their antitumor efficacy and safety in solid tumors with heterogeneous tumor populations and immunosuppressive TMEs, including glioblastoma, will be required. Ultimately, to be ideal off-the-shelf effector cells for the treatment of glioblastomas, NK-specific CAR iPSC NK cells will need to be produced in large quantities and modified to express cytokines that play a major role in stimulating NK cell expansion and cytotoxic functioning such as IL-15 [171,232], to relieve the immunosuppression mediated by TGF-β released from the TME [57], to express checkpoint inhibitors (as seen in CAR T cell therapy) [109], and to enable multi-specific targeting [220]. The advantages and the disadvantages of CAR NK cells close to off-the-shelf effector cells for glioblastoma therapy are summarized in Table 3.
COMBINATION THERAPY
Combination with conventional chemo-/radiotherapy
Lymphoid cells independently perform homeostatic regulation of resting and memory cells, so a rapid proliferation of remaining or infused lymphocytes occurs to recover normal lymphocyte numbers after periods of lymphopenia [59]. Because these homeostasis-induced T cells respond to tumor antigens at a lower dosage than naïve cells [26], the administration of TSAs in the form of a vaccine or ex vivo expanded adoptive T cell transfer during this recovery time can induce a disproportionate enhancement of effector cell populations that increases antitumor efficacy [96,181]. The induction of lymphodepletion in patients before T cell-based immunotherapy can be achieved using total body irradiation or non-myeloablative chemotherapy [157]. Another therapeutic advantage of lymphodepletion prior to immunotherapy is the ability to eliminate immunosuppressive cells, including myeloid derived suppressor cells (MDSCs) and Tregs [7]. Lymphodepletion before immunotherapy has been applied to various types of adoptive T cell therapy [176].
Conventional adjuvant therapies for patients with glioblastomas, such as radiotherapy and chemotherapy, are independently immunosuppressive [52]. However, they can also induce favorable immune responses by changing the TME to increase the antitumor efficacy of T cell therapy. In addition to cancer cell death caused by DNA damage, which triggers the release of danger signals, radiation can cause phenotypic changes in tumor cells that enhance tumor cell recognition and elimination, including the upregulation of MHC class I, NKG2D ligands, co-stimulatory receptor CD80, death receptor Fas, and intercellular adhesion molecule 1 [41,56,173]; the induction of proinflammatory cytokines such as TNF-α, IL-1β, IFN-γ, CXCL9, CXCL10 and CXCL16, which attracts T cells into the tumor [41,56]; and possibly the disruption of the tumor vasculature and the BBB, which would also increase T cell trafficking [179]. These immune-favorable responses in the TME can produce an endogenous and systemic antitumor immune response. The abscopal effect, an antitumor immune response that occurs outside the radiation field, suggests the potential for radiation-induced systemic antitumor immunity [56]. However, the rare occurrence of the abscopal response indicates that this endogenous immune response is generally weak. Thus, local radiation therapy can be harnessed in combination with immunotherapy to induce a potent systemic antitumor immune response. Combining CAR T cell therapy with radiotherapy has been shown to improve the antitumor efficacy of each monotherapy in preclinical models with a subset of solid tumors and glioblastoma [42,226]. In two independent syngeneic mouse models of glioblastoma, a sublethal dose of local radiotherapy combined with NKG2D CAR T cell therapy exerted synergistic activity by promoting the migration of CAR T cells to the tumor site, which increased the effector functions and prolonged survival [226]. Chemotherapeutic agents similar to local irradiation can also enhance the antitumor immunity of adoptively transferred T cells or CAR T cells via the upregulation of tumor antigens [78], elimination of immunosuppressive cells [253], and extension of cell survival [214].
Combination with immunotherapy
Immunotherapy, such as immune checkpoint inhibitors and oncolytic viruses, is another candidate for combination with CAR T cell therapy. Immune checkpoint inhibitors restore the activity of effector cells that can recognize and attack cancer cells. Despite clinical success in various cancers, including melanoma, non-small cell lung cancer, and renal cell carcinoma [203], anti-PD-1 monotherapy has not shown a significant survival benefit in patients with glioblastoma [177]. Immune checkpoint blockades that target the PD-1/PD-L1 and CTLA-4 pathways have been found to increase the activity of CAR T cells in preclinical studies of glioblastomas [194,245]. Two clinical trials of CAR T cell therapy combined with immune checkpoint inhibitors are currently progressing. One of them is a phase I clinical trial (NCT04003649) testing the safety and feasibility of L-13Rα2 CAR T cells administered alone or together with nivolumab (anti-PD-1) and ipilimumab (anti-CTLA-4) to patients with recurrent/refractory glioblastoma, and the other is a clinical study (NCT03726515) to assess the safety and tolerability of EGFRvIII CAR T cells in combination with pembrolizumab (PD-1 inhibitor) in patients with newly diagnosed EGFRvIII+, MGMT-unmethylated glioblastoma. However, systemic immune checkpoint inhibitors can block the checkpoints of Treg cells, as well as those of CAR T cells, which can enhance Treg cell function, as mentioned above.
Oncolytic viral infection and subsequent immunogenic tumor cell death can turn tumor cells into large-scale producers of tumor-specific neoantigens, cytokines, and chemokines, which converts the TME from immunosuppression to immune stimulation and thus augments T cell effector function and trafficking [86]. Oncolytic viruses can also induce the production of type I IFNs (IFN-αβ) in the TME to promote T cell proliferation, effector function, and immune memory function [37]. In addition, IFN-β can modulate the TME by inhibiting Treg cell activation and proliferation and disrupting tumor microvessels [255]. This potential for TME modulation suggests that oncolytic viruses and CAR T cell therapy could have synergistic antitumor effectiveness [3]. Combined therapy of B7-H3 CAR T cells and IL-7-loaded oncolytic adenoviruses has been shown to enhance T cell proliferation and reduce T cell apoptosis in vitro, and it produced a synergistic survival benefit in glioblastoma xenograft mouse models [74]. In another study of glioblastoma, a combination of oncolytic adenoviruses armed with CXCL11 and B7-H3 CAR T cells produced increased infiltration of the effector cells and decreased proportions of immunosuppressive cells such as MDSCs and Tregs in the TME. In animal models, glioblastoma that was not inhibited by B7-H3 CAR T cells alone was inhibited when CXCL11-armed oncolytic viruses were added [219]. In addition, a herpes simplex 1-based oncolytic virus expressing IL-15/IL-15Rα combined with EGFR CAR NK cells demonstrated increased synergistic antitumor effects and a significant survival gain in glioblastoma-bearing mice [122].
Combination with targeted therapy
Small-molecule inhibitors block intracellular signal transduction pathways such as tyrosine kinases and mitogen activated protein kinases in tumor cells, which deregulates cell proliferation and differentiation. Small-molecule tyrosine kinase inhibitors (TKIs) have been shown to have great antitumor efficacy in treating hematologic malignancies [191] and a variety of solid tumors [197]. Monotherapy with small-molecule TKIs, however, has displayed limited treatment outcomes in clinical studies of patients with glioblastoma [205]. Combining TKI therapy with CAR T cells in murine models produced synergistic antitumor effects in other types of solid tumors [107,235]. The combination of LB-100, a small-molecule inhibitor of protein phosphatase 2 A (involved in cell-to-cell adhesion), and CAIX-specific CAR T cells has been found to have synergistic antitumor effects in glioblastoma animal models [36]. These results suggest that TKIs could induce synergistic effects in combination with CAR T cell therapy for glioblastoma.
In a particularly useful innovation, CAR T cells can be designed to switch off by administering a small molecule that chemically disrupts a heterodimer [63]. CAR T cells incorporate a protease and CAR degradation moiety (degron) that can be switched on in the absence of the protease inhibitor asunaprevir. The degron is cleaved from the CAR by the protease, and it is switched off in the presence of asunaprevir; thus in the absence of the inhibitor, the degron is cleaved from the CAR by protease, leading to the degradation of the CAR [83]. These CAR T cells with a switch off function provide a controllable way to improve the safety of CAR T cell therapy and reduce the risk of CRS.
CONCLUSIONS
Glioblastoma has been an immunologically “cold” tumor characterized by a paucity of tumor infiltrating effector cells because it has high antigenic heterogeneity, a low mutational burden, an exceptionally immunosuppressive TME, and restricted immune access. Therefore, the clinical outcomes of immunotherapy for glioblastomas have been poor compared with those for other types of cancer. Nonetheless, cell transfer therapy using effector cells such as T and NK cells has been developed to overcome immune escape mechanisms and allow the cells to survive in the immunosuppressive TME.
The identification of patient-specific neoantigens derived from tumor mutations has expanded the usable repertoire of TSAs in glioblastoma. T and NK cells have also been engineered using modern genetic technologies to have multiple functionalities, including cytokine production, multiple antigen recognition, and trafficking enhancement with immune favorable modification of the TME through the inhibition of immunosuppressive molecules. BiTEs that facilitate optimal interactions between T cells and tumor cells can potentiate the cytotoxicity of effector cells. iPSC-derived NK receptor-specific CAR NK cells are close to ideal off-the-shelf effector cells. Further efforts will be needed to learn more about immune escape mechanisms and optimize effector cell functions using that knowledge. In addition, an effort is required to find potent combinatorial therapeutic strategies that enhance antitumor efficacy and minimize the toxic effects of T or NK cell therapy for glioblastoma.
 XML Download
XML Download